




精选内容
-
疾病科普 | 发作性运动诱发性运动障碍与16p11.2染色体微缺失
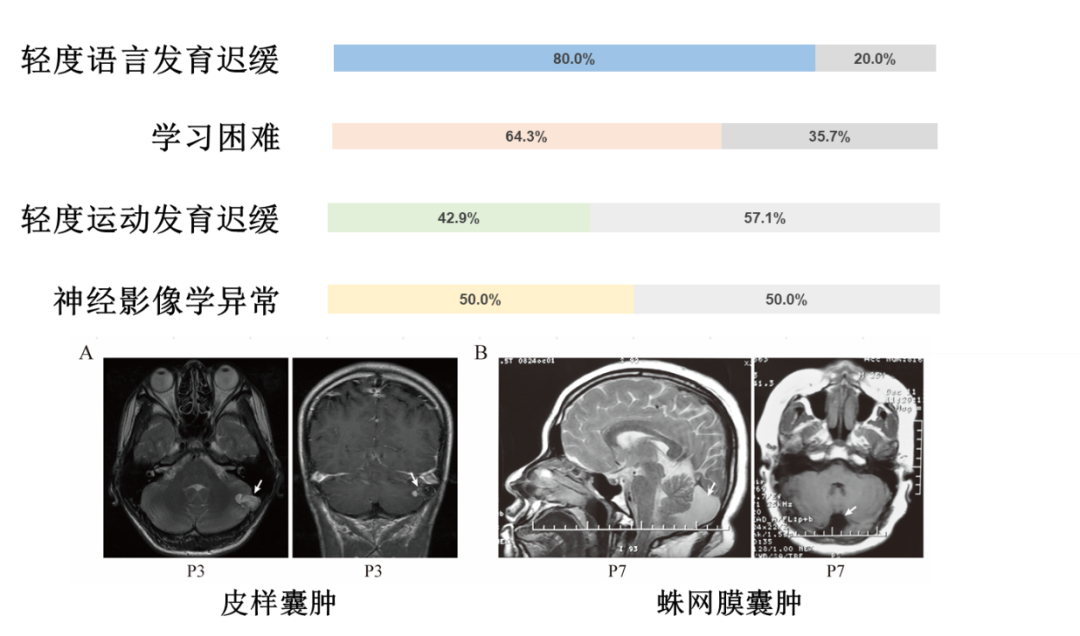
疾病科普|发作性运动诱发性运动障碍与16p11.2染色体微缺失什么是发作性运动诱发性运动障碍?发作性运动诱发性运动障碍,英文简称PKD,是最常见的发作性运动障碍疾病。PKD主要在儿童期和青春期起病,表
 耿鑫医生的科普号
耿鑫医生的科普号 2023年11月29日
2023年11月29日 2998
2998
 0
0
 0
0
-
脊髓固有性肌阵挛是运动神经疾病吗?
脊髓固有性肌阵挛(propriospinalmyoclonus,PSM)是突然的肌肉抽动,主要累及腹部、躯干和颈部肌肉,是一种罕见的运动障碍疾病。有症状性PSM、特发性PSM和功能性PSM。该病少见,临床上容易误诊而延误治疗。尤其是入睡期PSM很罕见,其肌阵挛常导致入睡困难,患者可能因此出现对睡眠的恐惧,出现焦虑和抑郁,也可自伤或伤他人。肌阵挛主要累及躯干肌肉,形式多变、强度变化无常。肌阵挛常始于胸腹部,顺着脊柱向上肢、下肢和颈部扩散。可往返反复发生,可单独出现,间隔期长短不一,也可呈丛集性发生,特发性主要发生在患者躺下时。入睡期PSM诊断:(1)主诉以颈部、腹部、躯干部位为主的突然抽动;(2)抽动发生于清醒放松状态,和思睡期,即患者试图入睡时;(3)抽动在思维活动时或进入稳定睡眠时消失;(4)抽动可导致入睡困难;(5)上述症状无法用其他睡眠障碍、神经系统或其他系统疾病、精神障碍以及药物使用解释。基于肌阵挛只发生在患者由清醒向睡眠或由睡眠向清醒转换的过程中这一现象,推测特定的意识状态可能对脊髓的运动产生有易化作用。功能性PSM诊断:(1)急性起病;(2)症状快速进展,可短期内自发缓解;(3)既往躯体化障碍;(4)无客观性症状,合并精神疾病;(5)抽动表现多样、不一致,发作时注意转移可短暂停止;(6)面部肌肉受累。但半数功能性PSM和症状性PSM均可急性起病,仅11%PSM患者的症状存在快速进展,因此急性起病、快速进展对于诊断功能性PSM意义有待进一步明确。皮质动作准备电位(Bereitschaftspotential,BP)是功能性PSM特有,反映了额中央皮质在肌阵挛发作前的神经元活动,提示为肌阵挛动作做准备,见于约63%功能性PSM患者。入睡期PSM常有腹部抽动主诉,与入睡明确相关,而症状性PSM常有明确原发病因及器质性病灶体征,诊断相对容易。功能性PSM则以发作形式多样、起源多灶为特点,常合并精神障碍,脑电图BP现象是特异性表现,但仅在2/3患者中出现,有时诊断较为困难,需综合考虑临床电生理表现。治疗症状性PSM首要是病因治疗。对症治疗主要为氯硝西泮(0.5~2mg每晚一次)等苯二氮卓类药物,德巴金、唑尼沙胺对部分患者有效。其他药物包括卡马西平、泼尼松、巴氯酚,疗效尚不明确。对神经电刺激以及认知行为治疗有效支持功能性PSM诊断。肉毒素治疗可用于治疗功能性PSM。左乙拉西坦可能有效。
 王祥瑞医生的科普号
王祥瑞医生的科普号 2022年10月09日324
0
12
2022年10月09日324
0
12
-
运动障碍可以发生在很多其他疾病中
运动障碍,并不是单纯的运动不能(瘫痪),而是一个很大的概念。它包括帕金森综合征、僵直综合征,还包括震颤、肌张力障碍、舞蹈症、投掷症、手足徐动症等许多症状。一般来说,目前主要认为运动障碍的症状来自大脑深部结构的损害,最为典型的损害部位是基底神经节。大脑深部损害的原因,可能是神经变性(神经细胞逐渐的死亡),也可能是结构性的损伤(如脑积水、脑内占位性病变等)。除此以外,还有很多全身性的疾病,也会表现为运动障碍的症状。全身性的疾病导致的运动障碍,最常见的原因是感染性疾病、自身免疫疾病、副肿瘤综合征、代谢和内分泌疾病等。这可以涉及很多专业、很多科室,因此有时候排查起来并不是那么容易。然而,要识别和治疗潜在的全身性疾病,对于病人来说是很重要的,这可以从根本上改善神经系统的症状。相当于是“治本”而不是“治标”了。这也是运动障碍疾病中能够“治本”的一个重要方向。以下列出有运动障碍症状,但是是全身性疾病的常见病因,供医生排查:感染性疾病●Whipple病:眼-咀嚼肌节律性运动● 神经梅毒:帕金森综合征,舞蹈症● 中枢神经系统结核:震颤、舞蹈症、肌阵挛、肌张力障碍、帕金森综合征● 人免疫缺陷病毒感染:偏身舞蹈症、震颤、帕金森综合征、肌张力障碍● 弓形虫病:偏身舞蹈-偏身投掷症● 脑囊虫病:(罕见)帕金森综合征、偏身舞蹈症● 莱姆病:帕金森综合征● 链球菌感染:帕金森综合征、小舞蹈病(儿童)● 其他病毒感染自身免疫性疾病● 系统性红斑狼疮:舞蹈症、帕金森综合征(少见)● 干燥综合征:帕金森综合征● 抗磷脂抗体综合症:(罕见)帕金森综合征、舞蹈症● 桥本脑病:震颤、共济失调、肌阵挛● 僵人综合症:脊柱前凸、共济失调● 神经白塞病:舞蹈症、共济失调● 乳糜泻:共济失调、帕金森综合征、舞蹈症副肿瘤疾病● 抗Yo抗体:共济失调、震颤● 抗NMDA受体抗体脑炎:肌张力障碍、口面运动障碍、投掷症、肌节律性运动● 抗Amphiphysin抗体:僵人综合征(脊柱前凸、肌肉强直、共济失调)● 抗Hu抗体:肌张力障碍、舞蹈症、震颤、帕金森综合征● 抗CV2抗体:舞蹈症、肌张力障碍、共济失调● 抗Ma1/Ma2抗体:帕金森综合征● 抗VGKC抗体:肌阵挛● 抗Tr抗体:共济失调● 抗Ri抗体:肌张力障碍、帕金森综合征(类似PSP)、斜视眼阵挛-肌阵挛● 抗VGCC抗体:共济失调代谢性疾病● 肝豆状核变性:肌张力障碍、共济失调、扑翼样震颤● 获得性肝脑变性:口颊舌运动障碍、帕金森综合征● 血色素沉着病:共济失调、震颤、帕金森综合征● 肾衰竭:姿势保持不能、不宁腿综合症、帕金森综合征(罕见)内分泌疾病● 非酮症性高血糖:偏侧舞蹈-偏身投掷征、姿势保持不能● 低血糖:阵发性舞蹈症● 甲状腺功能亢进症:震颤、舞蹈症● 甲状腺功能减退症:帕金森综合征● 甲状旁腺功能减退症:帕金森综合征、共济失调、震颤血液疾病● 红细胞增多症:舞蹈症● 舞蹈症-棘红细胞增多症:舞蹈症、进食性肌张力障碍●McLeod综合症:舞蹈症脂质体贮积症● 戈谢病:帕金森综合征、肌张力障碍中毒疾病● 一氧化碳:帕金森综合征● 镁帕金森综合征●MPTP:帕金森综合征● 麻黄碱:帕金森综合征● 二硫化碳:帕金森综合征● 氰化物:帕金森综合征、肌张力障碍、睁眼失用● 乙醇:共济失调、帕金森综合征● 铊:舞蹈症这样的列举,让我们对先前解释过的一类帕金森综合征,有更多的理解。一个简单的“感染”实际上背后还有很多可能性。帕金森综合征的病因参考资料:PoeweW,Djamshidian-TehraniA.Movementdisordersinsystemicdiseases.NeurolClin.2015Feb;33(1):269-97.
小光医生 2022年10月07日374
0
1
2022年10月07日374
0
1
-
那些可治的遗传性运动障碍:补充维生素
在罕见的累及神经系统的运动障碍疾病中,有很大一部分比例是遗传基因相关的,我们接下来分别列出它们。希望这对医生来说是一份学习的资源,对患者而言,有时候是一个继续明确诊断的理由。今天列举:通过补充维生素,来达到治疗效果的遗传性罕见运动障碍疾病:无β脂蛋白血症致病基因:甘油三酯微粒转运蛋白(MTTP)运动障碍表现:共济失调、舞蹈症、肌张力障碍、帕金森综合征其他临床表现:色素性视网膜炎、痴呆、癫痫发作、棘红细胞增多症、脂肪吸收障碍治疗方法:饮食限制脂肪、维生素E、维生素A治疗效果:早期治疗可预防症状发生芳香族氨基酸脱羧酶缺乏致病基因:芳香族氨基酸脱羧酶(AADC)运动障碍表现:肌张力障碍其他临床表现:发育迟缓、肌张力低下、动眼危象治疗方法:多巴胺激动剂、单胺氧化酶抑制剂、维生素B6治疗效果:部分患者可逆转症状维生素E缺乏共济失调致病基因:α-生育酚转移蛋白(TTPA)运动障碍表现:共济失调、肌张力障碍其他临床表现:周围神经病、视力受损治疗方法:维生素E治疗效果:早期治疗可预防症状生物素-硫胺素(维生素B1)反应性基底节病致病基因:硫胺素转运体(SLC19A3)运动障碍表现:发作性或进展性肌张力障碍、帕金森综合征、共济失调其他临床表现:脑病、癫痫发作(常因代谢代偿不足诱发)治疗方法:生物素+硫胺素,避免或治疗诱因治疗效果:早期治疗可以预防或逆转症状生物素(维生素H)酰胺酶缺乏致病基因:生物素酰胺酶(BTD)运动障碍表现:共济失调、痉挛性瘫痪、肌张力障碍其他临床表现:发育迟缓、癫痫发作、视觉和听觉损害、皮肤改变治疗方法:生物素治疗效果:早期治疗可预防症状脑叶酸缺乏致病基因:叶酸转运体(FLR1、SLC46A1)运动障碍表现:共济失调、肌张力障碍、肌肉痉挛其他临床表现:发育迟缓、癫痫发作、精神症状治疗方法:亚叶酸治疗效果:早期治疗可预防症状钴胺素(维生素B12)缺乏致病基因:钴胺素缺乏(多基因)运动障碍表现:共济失调、肌张力障碍、肌肉痉挛其他临床表现:发育迟缓、脑病、癫痫发作(常因代谢代偿不足诱发)治疗方法:钴胺素(维生素B12)衍生物治疗效果:早期治疗可减轻症状辅酶Q10缺乏致病基因:辅酶Q10缺乏(多基因)运动障碍表现:共济失调、肌张力障碍、震颤、肌肉痉挛其他临床表现:多种不同表现治疗方法:辅酶Q10治疗效果:可预防或逆转症状高胱胺酸尿症致病基因:胱硫醚β-合酶(CBS)运动障碍表现:肌张力障碍、帕金森综合征其他临床表现:发育迟缓、近视、异位晶状体、精神症状治疗方法:维生素B6、饮食限制甲硫氨酸、甜菜碱治疗效果:早期治疗可预防大部分症状 丙酮酸脱氢酶复合物缺乏症致病基因:丙酮酸脱氢酶复合体(多基因)运动障碍表现:慢性或发作性肌张力障碍、共济失调其他临床表现:发育迟缓、脑病、癫痫发作治疗方法:维生素B1、生酮饮食、三庚酸甘油酯治疗效果:可减轻症状参考资料:LynchDS,WadeC,PaivaARB,JohnN,KinsellaJA,MerwickÁ,AhmedRM,WarrenJD,MummeryCJ,SchottJM,FoxNC,HouldenH,AdamsME,DavagnanamI,MurphyE,ChatawayJ.Practicalapproachtothediagnosisofadult-onsetleukodystrophies:anupdatedguideinthegenomicera.JNeurolNeurosurgPsychiatry.2019May;90(5):543-554.
小光医生2022年08月15日239
0
0
-
发作性运动诱发性运动障碍临床表现和遗传学研究进展
发作性运动诱发性运动障碍(Paroxysmal Kinesigenic Dyskinesia, PKD)(MIM 128200)是发作性运动障碍(Paroxysmal dyskinesia)中的最常见的一种,是一组由突然运动所诱发的非随意运动障碍疾病,发作时以异常运动或异常姿势为特征,如肌张力障碍、舞蹈样动作、手足徐动、投掷样动作等,每次持续数秒至数十秒,发作间期正常1, 2。PKD是一种高度异质性疾病,大部分研究认为与遗传有关,随着分子生物学和遗传学技术的发展,该病的首个致病基因被发现3-5,由此对其的研究也在不断深入,本文将该疾病的临床表现及其遗传学的研究进展综述如下。 1.PKD临床特征 PKD由Kertesz于1967年首次报道2,2004年Bruno等提出了PKD的临床诊断标准1:由突然动作诱发;发作持续时间短暂(小于1分钟);发作期间意识清晰;发病年龄1-20岁,如有家族史,起病年龄可适当放宽;苯妥因钠或卡马西平能有效控制发作;神经系统体格检查及神经电生理检查正常且排除其他疾病。 PKD发病年龄可从6个月至33岁不等,但多见于7-15岁青少年,男女比例4:1~8:1,尤其在散发病例中,男女差异更为明显6。典型的PKD发作往往由突然动作诱发,比如起跑、起立开门或者接电话等,运动形式、速度、幅度的改变,意图动作甚至在持续动作中加入其他动作时也能诱发6-8,此外声音或图像的刺激、过度通气、情绪紧张等也可诱发6。70%的PKD患者发作前有预感,多数表现为受累肢体无力感1。部分患者在出现预感后可通过减慢患侧肢体动作以阻止发作6。PKD发作形式包括肌张力障碍、舞蹈样动作、投掷动作或以上非随意运动形式的任意组合6,其中肌张力障碍是最常见的发作形式7。通常发作多累及一侧肢体,也可两侧肢体同时受累或一侧重于另一侧。30%的患者发作时可累及面部肌肉,并因此出现言语表达障碍9。在频繁发作的患者中,发作间期存在“不应期”,即在前次发作后的约20分钟内,任何运动都不能再次诱发发作10。95%的患者发作时间小于1分钟1,对于发作持续时间过长的“PKD”患者,须考虑是否存在继发性因素,如心因性运动障碍等。值得注意的是,部分PKD患者还合并有婴儿惊厥(Infantile Convulsions,IC)、良性家族性婴儿惊厥(Benign Familial Infantile Seizures,BFIS)、婴儿惊厥伴阵发性舞蹈手足徐动症(Infantile Convulsions and Paroxysmal Choreoathetosis,ICCA)、偏头痛(migraine)、偏瘫型偏头痛(Hemiplegic migraine, HM)、发作性共济失调(Episodeic ataxia, EA)等发作性疾病4, 11-31。PKD患者每天发作频率各异,多数患者每日发作1-20次不等,也有患者每日发作20次以上1。PKD发作频率的高峰期往往出现在青春期,发作频率最高可至30-100次/天。PKD有自愈倾向,多数患者发作次数在20岁后逐渐减少,部分在30岁后甚至完全消失1。绝大多数患者对抗癫痫药物敏感,尤以卡马西平为著,研究表明86%的患者对卡马西平或苯妥英钠敏感1。在儿童PKD患者中,苯妥英钠用量与治疗癫痫时用量相似,而在成人患者中小剂量即可控制症状6。Houser等建议成人患者药物用量苯妥因钠每日5mg/kg或卡马西平每日7-15mg/kg9。托吡酯、巴比妥类药物亦可作为治疗药物6。由于PKD的自愈性,该疾病预后良好,对患者远期生存无影响1,但对于未确诊和治疗的病人,频繁发作会明显影响患者生活质量。 2.PKD遗传学研究 已报道的PKD病例中大部分呈家族性,也有散发病例。家族性PKD遗传方式多为常染色体显性遗传,伴有不完全外显,但也有隐性遗传的家系报道32-36。家族性PKD中,单纯性PKD较少见,多伴有IC、BFIS、ICCA、偏头痛或其他神经系统疾病。目前共发现了三个与PKD有关的位点EKD1(Episodic Kinesigenic Dyskinesia 1),EKD2,EKD336-38。 2.1 EKD1与PRRT2基因 对PKD致病基因定位研究的突破来源于对PKD与ICCA综合征同质性的认识35。ICCA综合征于1997年由Szepetowski等首次报道,该疾病患者主要表现在婴儿时期出现非热性惊厥和青少年期发生发作的性舞蹈手足徐动症(choreoathetosis,CA)39。家族性ICCA综合征亦呈常染色体显性遗传。连锁分析和单倍型分析将致病基因定位于16号染色体D16S401与D16S517之间约10cM的区域(16p12-q12)39。之后Tomita等于1999年对8个日本PKD家系进行连锁及单体型分析,将PKD基因定位于16p11.2-q12.1,并命名为EKD1(Episodic Kinesigenic Dyskinesia 1)38,该区域与之前的ICCA位点有6.6cM的重叠区域39, 40,这些家系具有典型的PKD发作特点,且其中部分患者有婴儿期非热性惊厥病史。由于交叉的临床表现及定位,故许多学者认为PKD与ICCA综合征可能是同一种疾病的不同临床表现。之后,Bennettn等对一个美国PKD家系的研究将并将致病位点定位于16p11.2- q12.1上的D16S3100和D16S771之间约18cM的区域内32,这个区域与先前日本家系的PKD位点有9.8cM的重叠,而与ICCA综合征的位点有3.4cM的重叠,这个结果更进一步证实了PKD和ICCA遗传同质性的观点。2011年,中国的研究小组率先证实PRRT2基因(Proline-rich transmembrane protein 2, PRRT2)为家族性PKD的致病基因3-5。 2.1.1 PRRT2突变与表型 PRRT2基因位于染色体16p11.2,包含4个外显子。自PRRT2基因被证实为家族性PKD的致病基因以来,迄今为止已有约20种PPRT2基因突变在PKD患者中得以明确41, 42,其中突变c.649dupC(p.R217PfsX7) 为突变热点,约占64%41-43。此外,由于PKD病人常合并其他神经系统发作性疾病,例如ICCA、BFIS等,且由于部分疾病的定位与EKD1接近,因此这些发作性疾病也引起学者的关注。近期的研究发现,在IC、BFIS、ICCA、HM、ET、热性惊厥(Febrile Seizures,FS)、偏头痛、发作性非运动源性运动障碍(Paroxysmal Non-kinesigenic Dyskinesia, PNKD)及发作性过度运动发性运动障碍(Paroxysmal Exercise-induced Dyskinesia, PED)家系及散发患者中也存在PRRT2基因突变4, 11-31, 41, 42, 44-53。这些结果提示上述一系列发作性疾病包括PKD有可能是同一疾病的不同表型。由于与PRRT2基因相关的疾病谱不断扩大,PRRT2-相关疾病(PRRT2-related Disorders,PRD)这一名称被提出41, 42,目前在PRD中发现的突变有57种,致病突变多位于PRRT2基因2号及3号外显子,其中无义突变占31种(54%),其他突变类型包括错义突变16种(33%)、剪切位突变6种(11%)和插入突变1中(2%)(表2)41, 42。而在所有突变中c.649dupC(p.R217PfsX8)出现的频率最高,由该突变导致的PRD患者占所有PRD患者的69%,该突变仍为突变热点(表2)41, 42。该突变为移码突变,其造成终止密码子提前出现,引起肽链截短,因此推测此突变导致PRRT2编码蛋白功能缺失进而引起疾病产生。目前PRRT2基因突变基因型与表型间的关系尚不明确,同一突变,例如c.649dupC,可导致几乎所有PRD表型3-5, 11-31, 41-44, 46-49, 52-58,其确切原因尚不得而知,但推测可能在于存在其他的突变引起修饰或甲基化形式的不同。 值得注意的是在目前已报道的与PRD病例中,具有阳性家族史的患者存在PRRT2基因突变的比例为88%,而散发病例只有35%存在PRRT2基因突变41, 42。其中对于PKD同样如此,PKD家系中发现PRRT2基因突变的占到91%,而散发病人仅有35%41, 42,并且散发PKD病人多为单纯性PKD,提示散发PKD的致病基因也许存在于EKD3(先前定位于EKD2的家系被证实存在PRRT2基因突变)14, 36, 37, 59。基于上述情况,对于存在阳性家族史的PRD患者,PRRT2基因筛查首选,在筛查策略上可优先筛查热点突变c.649dupC(p.R217PfsX7)。 2.1.2 PRRT2与PKD发病机制 PRRT2基因编码340个氨基酸,为跨膜蛋白,包含2个胞外区(氨基酸残基1-268及339-340),1个胞浆区(氨基酸残基290-317)和2个跨膜区(氨基酸残基269-289及318-338),且其跨膜区高度保守,提示具有重要的生理功能。PRRT2的mRNA在苍白球、小脑、丘脑底核、小脑脚、尾状核及大脑皮层均有高表达3。小鼠中PRRT2基因的表达量随年龄而有所不同,在胚胎期16天前小鼠脑组织中PRRT2基因表达量始终保持低水平而后逐渐上升,至出生后7日时在脑和脊髓大量表达,而到生后14日,PRRT2基因表达量达到最高峰,随后再次出现下降并且进入成年后继续维持下降趋势3。小鼠组织中PRRT2表达量随年龄而变化的现象符合PKD发病和缓解具有年龄依赖性的特征。 PRRT2基因突变中无义突变占到多数,且大部分位于N端的胞外区(表2),其结果是引起肽链不同程度的截短,并导致C端跨膜区的缺失。通过对这些截短蛋白的功能研究后证实截短的PRRT2蛋白不能锚定于胞膜,由此推测膜定位功能的缺失可能与疾病的发生有关3。而大部分错义突变位于含有跨膜区的C端(表2),这些点突变可能直接造成跨膜区功能的异常而同样导致膜定位功能的改变。体外培养的细胞系中截短的PRRT2蛋白并没有表达,这一现象推测可能与无义突变介导的mRNA降解 (Nonsense-mediated mRNA Decay, NMD) 有关18。NMD是一种mRNA水平的调控机制,其通过识别和降解含有提前终止密码子的转录产物阻止有潜在毒害作用的截短蛋白的表达60。在PKD中由于PRRT2 mRNA降解引起单倍体不足(haploinsufficiency)61同样可能造成PRRT2蛋白功能缺失进而导致疾病的发生。 然而PRRT2是如何进一步引发疾病的发生仍不明确,但PNKD的致病机制给了PRRT2功能研究一丝启示。PNKD同样是发作性运动障碍疾病中的一种,其临床表现和PKD有着一定的相似,这提示两者的致病机制同样可能存在共通之处。PNKD蛋白参与了神经突触调节过程,并且PNKD基因突变会干扰多巴胺能信号传导过程62。巧合的是,既往一项酵母双杂交筛选研究中曾发现,PRRT2与囊泡蛋白SNAP25(Synaptosomal-associated protein,25kDa)存在相互作用63。近期的研究也证实了PRRT2与SNAP25是相互作用蛋白18。SNAP25是Q-SNARE突触前蛋白,其与突触融合蛋白1(syntaxin-1)及突触泡蛋白-2(synaptobrevin2),也称为囊泡相关膜蛋白-2(vesicle-associated membrane protein-2,VAMP-2)共同组成胞吐复合体(SNAP receptors, SNARE)参与神经突触囊泡胞吐过程64。此外,SNAP25还参与电压门控的钙离子通道(Voltage-gated calcium channels,VGCC)的调控,其中包括与偏瘫型偏头痛相关的Cav2.1钙离子通道42。SNAP25蛋白在基底节区高表达且参与了突触前膜钙离子介导的胞吐过程。研究发现在兴奋的神经元中SNAP25能负向调控VGCC,而SNAP25突变的小鼠兴奋性较野生型明显增高,但通过抗癫痫药物能改善小鼠的兴奋状态65。提示PKD的致病机制可能与PNKD类似由于参与突触调控过程的蛋白功能异常引起,并继而引起神经元的高兴奋性。作为SNAP25相互作用蛋白的PRRT2发生功能缺失后很可能通过影响SNAP25介导的突触囊泡胞吐过程或VGCC调控导致疾病的发生。 2.2 EKD2 2000年,通过对一个印度PKD家系的研究,将致病基因位点定位于16q13-q22.1之间的15.8cM的区域。由于该位点和之前在日本家系中发现的位点和ICCA综合征的位点不同,故把它认定是PKD的第二个位点,命名为EKD236。该家系后证实存在PRRT2基因突变14, 36。 2.3 EKD3 2002年,Spacey等人37报道了一个英国PKD家系,该家系成员中PKD患者发病年龄6-13岁,且均为单纯性PKD,不伴有IC、BFIC、ICCA等其它神经系统疾病。通过连锁分析和单倍体分析,其疾病位点与之前两个PKD位点和ICCA综合征的位点不连锁,并且在16号染色体上没有找到相关连锁区域,因此认为存在第三个PKD位点,且该家系为单纯性PKD,故认为第三个位点与单纯性PKD相关37, 59。 综上,随着首个PKD致病基因的明确,对于PKD及其相关疾病的认识在不断加深,虽然目前PKD的致病机制仍不完全明了,但随着研究的不断深入,PKD临床症状复杂多样及遗传异质性之谜将解开。同时也将会给其他PKD致病基因的发现和基因产物的研究提供新的视野。
姚要兵医生的科普号 2020年06月07日6171
0
1
2020年06月07日6171
0
1
-
帕金森病药物治疗如何预防运动障碍并发症?
药物治疗帕金森病在3~5年的蜜月期后发现,左旋多巴不能持续有效,随着时间延长,作用时间越来越短,疗效越来越差。更为严重的弊端是药物引起的症状波动、异动症等运动障碍并发症。运动障碍并发症往往成为晚期帕金
王茂德医生的科普号 2019年06月12日1289
0
0
2019年06月12日1289
0
0
-
与癫痫发作相似的疾病-发作性运动障碍
1、发作性运动诱发的运动障碍(paroxysmal kinesigenic dyskinesia ,PKD)是最发作性运动障碍常见的发作类型,在儿童期或青年期起病,由突然的运动诱发,常出现在从坐位站起时,突然惊吓、过度换气也可诱发,表现由静止到运动时的发作,姿势肌张力不全或舞蹈综合征,一般持续时间为1-5分钟,每天又多次,发作时意识清,神经系统监察无异常,脑电图间期和发作期均为正常,65-72%有家族史,部分患者本人或家系成员有婴儿良性癫痫病史。2、发作性非运动诱发的运动障碍(paroxysmal non-kinesigenic dyskinesia PNKD),PNKD并不被突然的运动引起,可自发也可可由饮酒、咖啡、茶、疲劳、饥饿、精神刺激等诱发,发作时间教PKD长,常常持续5分钟以上,甚至数小时,发作频率较低,每天仅有1-3次,并可有数月的间隔期,可有感觉异常先兆,发作时语言功能受累,意识清,发病年龄小于PKD,PNKD可有家族史,但也可为散发病例。3、发作性持续运动诱发的运动障碍(paroxysmal exercise-induced dyskinesia PED),通常在持续运动后特别是行走和跑步后出现发作性的肌张力不全,多维持5-30分钟,停止诱发后才可缓解。4、发作性夜发性运动障碍(paroxysmal hypnogenicdyskinesia,PHD)PHD 睡眠期反复出现肌张力不全、舞蹈手足徐动样动作,发作不超过1分钟,每夜可发作多次。病因不明,抗癫痫药卡马西平对多数PHD疗效很好。PKD病人发作期未见异常放电。
王东明医生的科普号 2015年09月28日3742
1
1
2015年09月28日3742
1
1
-
随便聊聊:发作性动作诱发性运动障碍
随便聊聊(一):PKD2014-03-29caol聊聊神经遗传吧难得遇到一个相对空闲的周末,自然免不了和QQ群里的病友们聊聊,回答他们的一些咨询。今天聊的最多的是PKD群,群成员数从前几年的30多人已经壮大到269人,里面自然很多是我的老病人。讨论很热闹,大家不断爆料自己遇到的各种奇葩的就诊和生活工作经历,最让他们苦恼的是大部分医生对这个疾病的了解不够,在确诊之前经常被误诊;怕亲朋好友知道自己有这个病抬不起头来。他们想让我更多地宣传,让大家认识和关注这个疾病。想起2008年去河南山区采集PKD家系时,PKD致病基因还不明确,6年过去了,我们的团队一直致力于PKD诊治和研究工作,接触了很多PKD患者和家属,也深知他们内心的苦楚,也觉得有必要做些科普宣传。而且我们也感觉到PKD并没有预期的那么罕见。病例:女性,15岁,反复突然动作时诱发肢体扭动4年。2年前开始出现上课被点名回答问题,100米短跑起跑,听到电话铃响站立接电话等从静止状态到突然动作时出现右侧下肢的僵硬足内翻,右手不自主扭动,伴有面部肌肉的僵硬,讲话变音甚至讲话不能的情况。这种发作一般在迈出1-2步后出现,持续约10秒,但可长至30秒,发作时意识清晰,发作间期完全正常。发作前经常有要发作的预感,若此时停住不动,也常可控制避免不发。劳累时发作频繁,发作最多时每天可达50余次,少时只有数次,甚至一天不发。否认家族史。1岁时曾有一次发热惊厥史。查体未见异常。头颅MRI,脑电图等均正常。PRRT2基因检测发现c.649dupC杂合突变。诊断为:发作性运动诱发性运动障碍(Paroxysmal Kinesigenic Dyskinesia,PKD)诊断根据临床表现诊断一般不难,但需排除继发性PKD。辅助检查首选头颅磁共振及脑电图。(1)突然运动诱发的不自主运动;(2)每次发作一般不超过1分钟;(3)发作期无意识丧失,无疼痛感;(4)神经系统检查正常,排除其他疾病;(5)抗癫痫药物治疗有效,特别是苯妥英钠或卡马西平;(6)无家族史者发病年龄常在1-20岁。PKD对于卡马西平治疗效果很好。结合我们对许多PKD患者的治疗经验,我们推荐卡马西平为首选治疗药物,50毫克或100毫克,每天晚上睡前服用。仅极少数患者需要200毫克/天,,也有患者25毫克隔天服用达到很好的效果。如果有皮肤过敏等副作用,则需及时停药和换药。其他抗痫药物如苯妥英钠、拉莫三嗪、奥卡西平、妥吡酯等也能获得满意疗效。PRRT2基因检测可以进一步明确诊断,但一部分病人不存在PRRT2基因突变,目前认为与其它未知基因相关,有待于研究者们进一步明确。感谢本团队lancy,shirley等医生这几年为PKD的诊治做出的贡献。欢迎关注PKD病友群:PKD之家欢迎患友咨询:rjsn.sjyc@qq.com欢迎关注本微信平台:聊聊神经遗传吧
曹立医生的科普号 2014年03月30日6936
2
0
2014年03月30日6936
2
0
-
发作性运动障碍与癫痫
发作性运动障碍是否属于癫痫发作,目前专家观点不一致。一种观点认为:发作性运动障碍属于反射性癫痫、运动诱发的癫痫、家族性额叶癫痫,因癫痫与发作性运动障碍都具有反复发作性、刻板性、一过性、发作时间短、AED有效;而另一种观点认为:发作性运动障碍不同于癫痫发作,因脑电图发作期未见异常,临床发作不同于癫痫,无意识障碍、能回忆发病经过。 发作性运动障碍(paroxysmal dyskinesia, PD)是一类少见的发作性神经系统疾病, 表现为突然出现且反复发作的异常运动, 而发作间期表现正常。包括:发作性运动诱发性运动障碍( PKD)、发作性非运动诱发性运动障碍( PNKD)、发作性过度运动导致的运动障碍( PED)和睡眠诱发性发作性运动障碍( PHD)一、发作性运动诱发性运动障碍( paroxysmal kinesigenic dyskinesia, PKD): 亦称发作性运动诱发性舞蹈手足徐动症( paroxysmal kinesigenic choreathetosis, PKC ),分为原发性和继发性两种。原发性PKD 多有明确的家族史,呈常染色体显性遗传,基因定位16q11.2-q12.1;继发性PKD 可见于基底节梗死、多发性硬化、脑外伤、缺氧性脑病、甲亢、甲状旁腺减低引起的低血钙、糖尿病及颅内感染、颅内淋巴瘤、甲状腺功能减退等。 PKD 发病年龄为4 个月~ 57 岁,以儿童、青少年为主。临床以发作性姿势性肌张力障碍、舞蹈样动作及扭转样动作为特征,通常单侧或不对称,或者双侧呈交替性,可累及肢体、面部、颈部、躯干肌肉,严重可摔倒、说不出话。发作时意识清楚,无舌咬破、二便失禁,发作后无失忆或意识模糊。 诱因: 多由突然变化的运动或姿势所诱发,特别是经过一段时间的休息,如:从椅子上站立(开门、接电话、老师点名上台)、跑步(体育课、赶公交)、游泳、走路速度加快(迅速过人行道)、下车、下讲台、电梯。也可咀嚼、惊吓、紧张、热和冷诱发。持续时间数秒钟、数十秒钟,很少超过5min,发作频率高达100次/d, 通常每天发生,可以间隔1月或以上。许多患者主诉发作前有不同的异常感觉,称之为“先兆”。发作经常持续几秒到1~2min,一般不超过5min。 抗癫痫药物治疗有效。二、发作性非运动诱发性运动障碍 发作性非运动诱发性运动障碍 (paroxysmal nonkinesigenic dyskinesia, PNKD),即文献中报道的发作性肌张力障碍性舞蹈手足徐动 ( paroxysimal dyston ia choreoathetosis, PDC)。此型自发发生, 不被突然的运动诱发, 但发病年龄要早于PKD。 可被饮酒、咖啡、茶、月经、疲劳所诱发或自然发生, 但不被突然运动诱发,持续时间数分钟至数小时, 可超过1天,通常为5分钟到4小时。发作频率低,可每年数次到每天数次,起病时间中位数12岁(3-30岁),随年龄增加可部分或完全缓解,睡眠可终止发作。 发作表现肌张力障碍、徐动症、舞蹈症和芭蕾舞症的各种组合形式,可单侧或双侧,可局部或全身受累,语言功能常受到影响,发作期无意识丧失。发作间期神经系统检查正常,脑电图正常。家族史符合常染色体显性遗传,基因定位2q31-36。药物治疗效果不佳, 但氯硝西泮可能有效,Lee认为免疫机制方面起到一定的作用 MS 是继发性PNKD 的病因之一, 其它病因包括脑炎、围产期缺氧、胱氨酸尿症、甲旁低、假性甲旁低、甲亢、甲状腺毒症、TIA、头外伤、低血糖、基底节钙化、AIDS、糖尿病等。 PNKD 很难治疗, 抗惊厥药物反应欠佳。抗毒蕈碱的药物、苯甲托品、敏使朗等合用苯妥英钠有效,卡马西平、苯二氮卓 类药物、乙酰唑胺、苯海索和氟哌啶醇已用于试验治疗取得不同结果。左旋多巴反应不一。三、发作性过度运动导致的运动障碍( PED) 发作性过度运动导致的运动障碍 (paroxysimal exercise- induced dystonia, PED),亦称发作性持续运动诱发的舞蹈徐动症(paroxysmal exercise-induced choreoathethosis),在长时间运动后发生,发作前5-15min的走路或跑步。被动的肢体运动、讲话、咀嚼、应激、热冷、月经期、酒精可促发。发作频率每天1-2次到每月5次,持续5-30min。多累积双侧下肢,或单侧面部、颈部和躯干,但上肢少见。发病年龄24个月到30岁,多在儿童期,男女平等。常染色体显性遗传,基因定位于16q-11.2. 抗癫痫药物无效, 左旋多巴和乙酰唑胺可能部分有效。四、睡眠诱发性发作性运动障碍( PHD) 睡眠诱发性发作性运动障碍(paroxysmal hypnogenic dyskinesia, PHD ),亦称夜间发作性肌张力障碍(nocturnal paroxysmal dystonia, NPD),是在NREM 睡眠期反复出现的刻板的肌张力障碍或运动障碍(如投掷样或手足徐动、舞蹈样动作) 的发作。与额叶癫痫相混淆,多数认为属于常染色体显性遗传夜发性额叶癫痫。目前仍存有争议,一种观点认为本病类似于异常睡眠中的夜惊, 但短暂的持续时间、刻板性、反复发作性, 对卡马西平有效而对苯二氮卓类药物无效不支持这种观点。另一种观点认为本病是与睡眠有关的癫痫的一种表现形式。有人则认为本病与发作性夜惊、发作性夜游症构成了夜间额叶癫痫, 只是持续时间不同。 发病年龄从婴儿时期到50岁, 男女无差异,短时间发作持续时间为15~60s,长时间发作持续时间可达60min。短时间PHD发作持续时间一般不超过1min,发作通常在临床和脑电图觉醒之前,每晚可反复发作数次, 最多达15次, 每夜或几乎每夜均有发作,发作时眼睛张开, 头抬起, 随后出现躯体和肢体肌张力障碍的姿势, 伴随着投掷样或舞蹈样或手足徐动样的动作, 刻板, 常伴有声音出现,发作结束时神志清醒,如果不被打扰,可恢复睡眠。长时间PHD发作同短时间NPD类似,但持续时间长,可达1小时发作可致严重的睡眠障碍, 患者常有失眠的主诉。 对短时间发作者用小剂量卡马西平有良好的治疗效果, 睡前口服200mg, 如无效可逐渐增加剂量直到症状控制。对发作持续2min以上者无有效的处理方法
王爱华医生的科普号 2012年09月15日11532
2
0
2012年09月15日11532
2
0
-
发作性运动诱发性肌张力障碍- PKD
发作性运动诱发性运动障碍(Paroxysmal Kinesigenic Dyskinesia,PKD),过去也称发作性运动诱发性舞蹈样手足徐动症,是发作性运动障碍中最常见的类型,临床上以突然运动诱发的运动或姿势异常为特征,如短暂频繁发作的肌张力障碍、手足徐动、舞蹈样动作、投掷样动作等。由于该病少见,发作间期正常,容易被误诊。1、病因:PKD可分为原发性和继发性。原发性大多有明确的家族史,符合常染色体显性遗传方式,有外显不全现象,也有散发病例,其致病基因被定位于第16号染色体上,目前明确的PKD1型是由于PRRT2基因所致,其中c.649dupC突变是中国PKD1的突变热点,大约在60%左右。结合我们瑞金医院检测的结果,我们发现许多家系中有不完全外显的情况,父母之一携带突变但没有发病,传给了小孩,小孩发病了,由于现在很多是独生子女,这样就以散发的形式出现,但事实上还是遗传所致。继发性PKD见于多发性硬化、脑外伤、脑血管疾病、围产期缺氧性脑病及甲状腺功能亢进、糖尿病,低钙血症等代谢异常疾病。2、临床特点:PKD多在青少年期发病,男性居多,主要在突然改变姿势、运动方向或力量负荷时发生,静止状态下突发的自主动作是本病最重要的诱发因素,如坐位突然站立时,久站后跑步,转身、举手等,也可由紧张、惊吓、焦虑、兴奋、过度换气等非特异性因素诱发,约70%的病人主诉发作前有“先兆”,如受累部位肢体发麻、发凉、紧箍感、沉重感等。发作时患者无意识障碍,主要表现为肢体的肌张力不全、舞蹈、手足徐动、投掷样动作等,多为单侧肢体发作,部分患者累及双侧,少数呈左右交替发作,当面部或下颌肌肉受累时,可影响语言以及出现面部的奇怪表情等。发作持续数秒至数10秒,一般不超过5分钟。PKD发作频繁,许多病人每天发作近20次,尤其在青春期可高达30~100次,在20岁后可逐渐减少。3、诊断:根据临床表现诊断一般不难,但需排除继发性PKD。辅助检查首选头颅磁共振及脑电图。Bruno等人于2004年通过综合分析121例病人临床资料后提出最新的PKD诊断标准:(1)突然运动诱发的不自主运动;(2)每次发作一般不超过1分钟;(3)发作期无意识丧失,无疼痛感;(4)神经系统检查正常,排除其他疾病;(5)抗癫痫药物治疗有效,特别是苯妥英钠或卡马西平;(6)无家族史者发病年龄常在1~20岁。4、鉴别诊断:(1)发作性非运动诱发性运动障碍(Paroxysmal Nonkinesigenic Dyskinesia,PNKD):由多种因素诱发,如咖啡、酒精、疲劳等,而并非由突然运动诱发,症状与PKD相似,持续时间可以数分钟到数小时,但发作频率较低,每天仅有1~3次,可有数月的间隔期。MR-1基因是其致病基因。(2)发作性过度运动导致的运动障碍(Paroxysmal Exertion induced Dyskinesia,PED):症状发生与长时间或过量的运动有关,如走路、跑步等,最常累及脚部的运动,抗癫痫药基本无效。(3)阵发性共济失调(Episodic Ataxia,EA):可被突然的运动刺激触发,但主要表现为运动协调功能和平衡功能的障碍,常伴有头和手的姿势性震颤以及面部和手部肌肉的细抽搐。5、治疗:一般PKD对于抗癫痫药物治疗反应较好,特别是卡马西平和苯妥英钠。结合我们对许多PKD患者的治疗经验,我们推荐的首选治疗:得理多(卡马西平)50毫克至100毫克,每天晚上睡前服用。部分病人需要200毫克,如果有皮肤过敏等副作用,则需要换药。其他抗痫药物如拉莫三嗪、奥卡西平、妥吡酯等也能获得满意疗效。6、基因检测:有患者需要明确诊断,基因检测的话,可以和我们联系。【联系方式】邮箱:rjsn.sjyc@qq.com
曹立医生的科普号2011年12月08日40849
19
1
相关科普号
王茂德医生的科普号
王茂德 主任医师
西安交通大学第一附属医院
神经外科
2511粉丝397.1万阅读

秦兵医生的科普号
秦兵 主任医师
暨南大学附属第一医院
癫痫中心
5088粉丝34.7万阅读

张歆博医生的科普号
张歆博 医师
解放军联勤保障部队第903医院
神经内科
197粉丝151阅读
-

 推荐热度5.0马翔宇 主任医师山东大学齐鲁医院 神经外科
推荐热度5.0马翔宇 主任医师山东大学齐鲁医院 神经外科帕金森 280票
面肌痉挛 125票
三叉神经痛 74票
擅长:脑深部电刺激术(DBS,脑起搏器)手术治疗帕金森病、梅杰综合征、痉挛性斜颈、特发性震颤、肌张力障碍。率先提出标准化操作流程(SOP)指导下加速康复外科(ERAS)脑起搏器手术,团队目标:精准高效,又快又好!神经内镜手术微创治疗三叉神经痛及面肌痉挛。 -

 推荐热度5.0李建宇 主任医师宣武医院 功能神经外科
推荐热度5.0李建宇 主任医师宣武医院 功能神经外科锥体外系疾病 80票
帕金森 73票
面肌痉挛 32票
擅长:帕金森病、面肌痉挛、肌张力障碍、特发性震颤、痉挛性斜颈、梅杰综合征、三叉神经痛、癫痫、抽动秽语综合症、颈肩痛、腰腿痛、精神外科 -
 推荐热度4.3李卫国 主任医师山东大学齐鲁医院 神经外科
推荐热度4.3李卫国 主任医师山东大学齐鲁医院 神经外科帕金森 173票
面肌痉挛 32票
锥体外系疾病 22票
擅长:专长:帕金森病、特发性震颤的手术治疗;痉挛性斜颈、梅杰(MEIGE)综合症、扭转痉挛等肌张力障碍、药物难治性癫痫、三叉神经痛、面肌痉挛的微创治疗; 神经外科常见病、多发病如:脑、椎管内肿瘤;