




精选内容
-
PW综合征介入硬化治疗病例
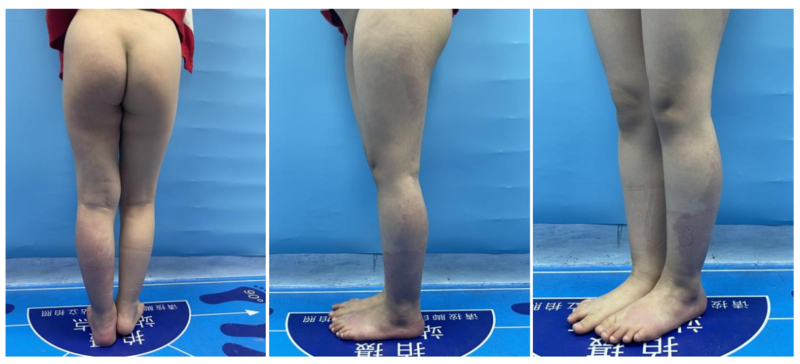
作者:狄奇申刚【基本信息】女、11岁【疾病类型】pw综合征【就诊时间】2022-11【治疗医院】首都儿科研究所附属儿童医院(三甲医院)【治疗方案】pw综合征微创介入治疗【治疗周期】3天【治疗效果】症状改善【推荐理由】罕见疾病,长期慢性病程,症状减轻。2022年11月,患儿因为左侧下肢可见红斑,并且皮肤温度也比对侧增高。在父母的陪同下来到首都儿科研究所附属医院的介入血管瘤科门诊,孩子左侧下肢肿胀增粗伴有红斑,最开始家长以为是胎记,没太在意,后来红斑没有好转,并且左下肢也增粗了,明显比对侧粗了1圈。赶紧到首都儿科研究所就诊。完善检查后提示是脉管畸形,在门诊医师详细的查体和问诊以后,得出诊断为pws综合征。在妥善安排好床位和手术日期,患儿完善术前检查后入院,第二天就安排入介入室治疗。孩子全麻下行血管腔内药物灌注。手术中就可以看到微动静脉瘘消失。手术时间不长,回病房后患儿卧床8小时,第二天就可以下床,下肢无肿胀,无压痛,无水泡坏死,下地活动良好。术后给予患儿口服免疫调节剂,延缓PW综合征脉管畸形进展,改善症状,皮温有所降低,顺利出院。科室简介首都儿科研究所附属儿童医院“介入血管瘤科”是北京首个儿童介入/血管瘤专科,一级临床科室,填补京津冀地区儿童介入治疗的空白。目前科室已开展儿童血管瘤、肿瘤综合介入、非血管介入及神经介入手术和治疗。本科室在儿童血管瘤、血管畸形、淋巴管瘤、淋巴管畸形;KT综合征、PW综合征;视网膜母细胞瘤、肝母细胞瘤、肝血管瘤、儿童肿瘤的介入综合治疗;儿童血管性疾病,如肾动脉狭窄、布-加氏综合征、急慢性动静脉血栓、血管狭窄、动脉炎;儿童脑血管疾病疾病的诊断和治疗都有着深入的研究。科室目前配备血管瘤、淋巴管瘤及介入医学门诊、病房、介入导管室、麻醉恢复室等完整的微创介入诊疗体系。病房目前开放12张床位,引进国际先进的西门子双C壁造影机、VbeamPerfacta595脉冲染料激光、Medtronic血管腔内射频消融导管、TEVASON便携式超声彩色多普勒诊断仪等先进介入诊疗设备,配备200余种儿科专用介入器材。拥有完善的消毒设备、国内领先的科室信息化管理系统,以及现代化的硬件配置,不仅保证医疗质量的提高,同时也为科室的进一步发展提供了强有力的支持。科室创始人申刚主任,从事血管瘤及儿科介入临床诊疗工作20余年,迄今为止主刀及参与各类介入手术近万余台,并携该领域杰出代表狄奇医生、李三林医生,充分利用新兴介入技术优势,弥补传统治疗不足,为患儿提供更加简便、安全、高效、微创、精准、并发症少的诊疗服务,为进一步完善我院疾病诊疗的综合能力,促进儿科事业的发展。
 申刚医生的科普号
申刚医生的科普号 2022年12月04日
2022年12月04日 264
264
 0
0
 0
0
-
60.早期使用生长激素治疗普拉德-威利综合征有什么好处?
国内外研究表明,生长激素是治疗小胖威利综合征的安全有效药物。生长激素可以促进小胖威利患者骨整生长,提高生长速率;可以帮助患者加速脂肪分解,减少脂肪堆积,增加肌肉质量及力量等。尽管目前国际上对PWS开始使用rhGH治疗的年龄尚未达成确切共识,根据以往的研究结果和治疗建议,GH起始治疗的时间通常在患儿出现肥胖前即2岁左右,早期治疗有利于改善身体状态。美国食品药品监督管理局(PDA)已经把小胖威利综合征纳入生长激素的综合征。
王强医生的科普号 2022年11月19日90
0
0
2022年11月19日90
0
0
-
KT与PW的鉴别是什么

 申刚医生的科普号2022年11月12日93
0
0
申刚医生的科普号2022年11月12日93
0
0
-
孩子3岁,pw综合症,盆腔和左腿到脚底都有,很担心
申刚医生的科普号2022年11月12日46
0
0
-
pw和kt什么区别呢?怎么区分
 尹杰医生的科普号
尹杰医生的科普号 2022年11月05日79
0
0
2022年11月05日79
0
0
-
狄医生您好!孩子患PW综合症,做了三次手术了,颜色和皮温都控制的很好,但是长度还在发展怎么办很着急
 狄奇医生科普号
狄奇医生科普号 2022年11月04日61
0
0
2022年11月04日61
0
0
-
我儿子一个腿粗一个细,做的CT左下肢较对侧增粗以皮下脂肪增多为著。左侧骼总动脉起始段较细,
申刚医生的科普号2022年10月31日38
0
0
-
PW综合征做完手术后颜色比之前深了正常吗?
申刚医生的科普号2022年10月31日51
0
0
-
Prader-willi综合征,小胖威利综合征,普拉德-威利综合征,附诊疗机构和组织
Prader-willi综合征(Prader-willisyndrome,PWS)也叫普拉德-威利综合征、小胖威利综合征,是一种罕见的、涉及基因印记的遗传性疾病。1956年由AndreaPrader,
付朝杰医生的科普号 2022年07月15日734
0
0
2022年07月15日734
0
0
-
认识普拉德-威利综合征
普拉德-威利综合征(Prader-WilliSyndrome),简称PWS,俗称小胖威利,是一种罕见的先天性疾病,因第15号染色体长臂(位置15q11-q13)遗传异常导致的终身性非孟德尔遗传的表观遗传性疾病,是多系统化异常的复杂综合征。此病会造成低肌张力、性腺功能减退、智力障碍、行为问题及过度摄食造成过度肥胖。尚无办法彻底根治。一、临床症状及特征临床症状复杂,不同年龄段表现不同,症状和严重程度个体差异大。1.新生儿及婴儿期:孕期胎动少、出生时体重偏低、肌张力低下(身体软)、喂养困难(不吃、吸吮和吞咽困难,常需鼻胃管灌食)、哭声微弱(不哭)、四肢活动力差(不动)、生长缓慢、嗜睡、反复呼吸道感染、肺换气不足、肺炎、睡眠窒息、喉头软化症及先天性心脏病等问题。PWS易诊断为脑瘫、脊髓性肌萎缩症(SMA)、重症肌无力等,早发现早干预治疗效果好。2.特殊外观(伴或不伴):窄脸、前额窄、长颅、单眼皮、杏仁眼、斜视、窄鼻梁、薄上唇、嘴角下垂、小嘴、身材矮小、皮肤白、发色较淡、小颌畸形、小手小脚、手狭窄且尺侧边缘较直、隐睾。3.食欲问题:患者无饱腹感并于1岁至6岁出现食欲亢进且无法自控,加之患者的新陈代谢率低,热量消耗慢,造成体重急速增长。过度肥胖将导致各种并发症:代谢紊乱、糖尿病、高血压、冠心病、脑卒中、非酒精脂肪肝、睡眠紊乱、睡眠呼吸暂停(嗜睡/打鼾)、呼吸道梗阻等,甚至猝死。严重过度摄食可导致胃肠道穿孔。4.运动发展:动作发展迟缓,如8个月才会抬头,1岁才会坐,2岁才会走,学龄儿童动作发展相比同龄人晚一到两年。虽随年龄增加,肌张力有所改善,但肌力、协调度、平衡感的缺失仍持续存在。5.智力/语言:轻度到中度智障,IQ平均70,少部份人呈严重智障或正常智力。语言发展迟缓(如3岁才会说话):构音缺陷、语言清晰度欠佳、语言重复。6.学习问题:高层次的抽象思维、数学计算能力、系统与次序性整合、听觉讯息能力差导致学习困难,造成生活技能、问题解决力低且社会能力差。但记忆力、阅读、尤其视觉认知、空间-概念组织能力较佳(如擅长拼图类游戏);语言理解尚可但表浅、语言表达能力不错,学习方式上以示范动作加上口语解释,可加强学习效果。7.情绪行为问题:挫败耐受差、情绪不稳、冲动、易怒、抠抓(自损)皮肤、程序化行为、固执、不合作、爱争辩、对立、违抗、藏食物、占有欲强、自尊心强、人际关系退缩、自言自语、多动症、强迫症、忧郁症(青少年以后可严重)、甚至暴力行为。8.性腺发育不良:性腺荷尔蒙通常不够,小男婴睾丸未降(单或双侧隐睾需手术)、阴茎短小;女生阴唇与阴蒂发育不良,青春期大多迟缓,未有生育报告。9.眼睛问题:集合性斜视/内斜视、近视、远视、散光、外眼角上斜、蓝巩膜、虹膜有Brush-Field(灰白)斑点、白内障。10.牙齿问题:因牙齿珐琅质太软、唾液黏稠、磨牙、反刍等,患者大多存在蛀牙、牙齿缺损、齿列异常等问题。11.体温调节异常:婴儿期体温不稳定常持续发热,年长儿及成年人体温敏感性改变。12.骨骼系统:脊柱侧弯发生率高,骨质疏松(易骨折)、髋关节发育不良(易脱位)、足外翻、下肢平衡异常等。需定期复查。13.其他:高疼痛阀值,对疼痛刺激相对不敏感。胃轻瘫:胃动力不足导致的胃排空延迟现象。甲状腺功能减退、肾上腺功能低下、皮肤瘙痒、麻醉后复苏(PWS患者可能对常规剂量的药物及麻醉剂出现异常反应需密切监控)、遗尿。偶发抽搐、癫痫或精神疾病。二、病因和发病机制15q11-q13区域异常导致PWS表型的机制尚不明了,可能与下丘脑功能紊乱或新陈代谢障碍有关。现研究显示15q11-q13区域存在SNRPN、NDN、MAGEL2、MKRN3和Cl5orf2印记基因,它们仅存于父源15号染色体的等位基因上。SNRPN位于印迹中心区域,可编码5种snoRNAs,被认为与PWS表型有密切关系,也是最可靠的诊断位点。正常人母源性15q11-q13区域SNRPN的CpG岛高度甲基化,而父源SNRPN的CpG岛未甲基化,因此母源性基因失活,而父源性SNRPN基因有表达(遗传印记)。当父源性15q11-q13区域SNRPN缺失(或功能缺陷)时,患儿即表现出PWS表型。PWS的致病原因及遗传机制非常特殊且复杂,绝大多数病例为新发突变,即父母亲皆正常,仅有极少数为遗传。PWS肇因于第15号染色体印迹基因区的基因缺陷,此基因缺陷来自父亲,或同时拥有两条来自母亲的带有此缺陷的第15号染色体,若此基因缺陷来自母亲,则导致Angelman'ssyndrome(天使综合症)。主要四种类型1、父源缺失型(deletion):绝大部分是因父亲第15号染色体15q11-q13区域有微小缺失,该区域涉及5-6Mb,根据近端断裂点不同分为I型和II型缺失。2、母源单亲二倍体(maternaluniparentaldisomy,简称UPD):20~25%是因两条第15号染色体都来自母亲。主要是由于卵细胞减数分裂时染色体不分离所致。3、印记中心突变或微缺失(IC):2~5%是因父源染色体15q11-q13 关键区域发生基因突变。4、平衡易位 (balancedtranslocation)或异常:约占1%因15号染色体与其他染色体发生不平衡结构重排所致。三、诊断(1)分子遗传学诊断方法1、高分辨率染色体分析(HRB):可检测染色体区域15q11-13的缺失 (deletion),大约可诊断70%患者。适用于大的缺失或染色体异常,无法检出UPD或印记基因突变、以及较小微缺失;2、荧光原位杂交法(FISH):利用位于15q11-13的探针,可检测出微细缺失。但无法检出UPD或印记基因突变;3、甲基化-特异性聚合酶链反应(MS-PCR):可以检出99%的PWS患者,但无法确定为哪一种遗传类型。另外1%由于是单碱基变异或者微小缺失(印迹突变和平衡易位)而无法检测出;4、甲基化特异性多重连接依赖性探针扩增法(MS-MLPA):能快速检测多种疾病相关基因的缺失或是重复突变,能够诊断99%以上PWS患者,能够区分缺失型和UPD;5、染色体微阵列分析(CMA):能在全基因组水平进行扫描,可检测染色体不平衡的拷贝数变异(CotyNumberVariant,CNV),仅能检测出该致病区域的缺失(75~80%),且不能区分缺失来自父源或母源。因此,即使做了微阵列检测也应同时行MS-PCR或MS-MLPA确定染色体突变的亲源性,两者的诊断率高达99%。此外,还可使用全外显子测序、全基因组SNP微阵列芯片、Southern印迹杂交、DNA甲基化(MSP)、AffymetrixCytoScan芯片。(2)疾病诊断目前国际上通行的PWS临床评分标准主要根据Holm等于1993年提出、2012年Cassidy等修正后的标准:包括6条主要标准、11条次要标准和8条支持证据。年龄<3岁总评分5分以上,主要诊断标准达4分即可诊断;年龄≥3岁总评分8分以上,主要诊断标准达5分即可诊断。四、治疗PWS的治疗应采用包括内分泌遗传代谢科、口腔科、新生儿科、眼科、骨科、康复科、神经内科、心理科、营养科等在内的多学科综合管理模式,根据不同年龄段患儿的表型特征,针对不同问题进行有效干预。1、肥胖控制:管住嘴,迈开腿。食欲异常是因下丘脑的功能异常所致,几近强迫性的摄食习惯通常会在6岁之前发生。应采取均衡、低热量的饮食,避免高糖、高脂肪食物,建立良好的饮食习惯及保持经常性的运动;2、生长激素(GH)治疗:可提高生长速率、减少脂肪堆积、增加肌肉质量及力量、促进热量消耗、促蛋白质合成、骨质密度增加等。生长激素治疗的患者体组织的改变因人而异,通常在治疗的第一年改变最大。应由医师评估患者情况(包括睡眠评估等),再考虑是否使用生长激素。生长激素引起的副作用,尚在观察及研究中,需注意的相关问题:①.脊柱侧弯:PWS患者脊柱侧弯发生病高,使用GH时,可能会因快速发育而加重脊柱侧弯,因此在GH治疗之前和治疗之后每6-12个月进行脊柱正侧位摄片检查。②.胰岛素抵抗与糖尿病:PWS患儿胰岛素水平可显著升高,对于肥胖的PWS患者可能会增加2型糖尿病的患病机率,因此PWS患者需要3-6个月定期监测糖脂代谢相关指标。③.阻塞性睡眠呼吸暂停(OSA):PWS儿童青少年OSA自然发生率为44~100%,GH治疗可能增大舌体和腺体的体积,减小本来就狭小的气道,可能加重OSA,导致患儿上呼吸道感染时发生猝死。美国药管局认为严重肥胖、严重呼吸问题和糖尿病的PWS患者不建议应用生长激素。3.其他内分泌激素治疗:男童隐睾和外生殖器发育不良可以应用睾酮及HCG治疗,疗效不佳建议2岁内手术治疗。甲状腺功能低下、皮质醇减低需要对症治疗。4.需于遗传咨询门诊定期追踪,家庭支持与遗传咨询是必要的,患者需要稳定而习惯化的环境。五、遗传咨询PWS的再发风险印记基因缺陷的类型有关,父源缺失和母源UPD为散发,父母基因均正常,再发风险约为1%。印记基因突变:如果父亲也是PWS病人,再发风险高达50%,如果父亲不是PWS病人,再发率很低。由于胎盘绒毛等组织的低甲基化状态,因此不推荐将其用于产前诊断;如确实需要产前诊断,可以在孕16-20周通过羊水脱落细胞DNA甲基化分析行产前诊断。PWS患儿鲜有生育的报道,其子代患PWS的概率与先证者的遗传机制及性别有关。理论上女性缺失型患者的子代有50%发生Angelman综合征的风险,而男性缺失型患者的子代有50%发生PWS的风险,故一般不建议PWS患者生育。
孙妍医生的科普号 2022年07月01日1262
0
0
2022年07月01日1262
0
0
相关科普号
申刚医生的科普号
申刚 主任医师
首都儿科研究所附属儿童医院
介入血管瘤科
1.8万粉丝30.5万阅读

张惠文医生的科普号
张惠文 主任医师
上海交通大学医学院附属新华医院
儿童内分泌遗传科
2461粉丝20万阅读

张沛医生的科普号
张沛 主治医师
商丘市第一人民医院
儿科
36粉丝25.9万阅读
-
 推荐热度5.0万平 主治医师上海交通大学医学院附属仁济医院(东院) 肝脏外科
推荐热度5.0万平 主治医师上海交通大学医学院附属仁济医院(东院) 肝脏外科肝移植 82票
肝母细胞瘤 13票
遗传代谢病 8票
擅长:小儿肝病及罕见病的临床诊治,尤其是儿童遗传代谢病、胆道闭锁、胆汁淤积症、肝母细胞瘤等疾病的肝移植手术治疗,包括酪氨酸血症、尿素循环障碍(如鸟氨酸氨甲酰转移酶缺陷、HHH综合症、瓜氨酸血症、氨甲酰磷酸合成酶缺陷、精氨酸血症、精氨酰琥珀酸合成酶缺陷等)、甲基丙二酸血症、丙酸血症、糖原累积症、原发性高草酸尿症、戈谢病、尼曼匹克病、家族性高胆固醇血症、Caroli病、枫糖尿病、肝豆状核变性、进行性家族性肝内胆汁淤积症、Crigler-Najjar综合症、线粒体病等。 -
 推荐热度4.5杨艳玲 主任医师北京大学第一医院 小儿神经内科
推荐热度4.5杨艳玲 主任医师北京大学第一医院 小儿神经内科遗传代谢病 3票
肌病 1票
发育迟缓 1票
擅长:遗传代谢性疾病的诊断与治疗 -
 推荐热度4.5商晓红 主任医师山东省立医院 小儿内分泌科
推荐热度4.5商晓红 主任医师山东省立医院 小儿内分泌科遗传代谢病 5票
佝偻病 3票
小儿糖尿病 3票
擅长:儿童矮小症、性早熟、糖尿病、先天性肾上腺皮质增生症、各类佝偻病等内分泌疾病;甲基丙二酸血症、戊二酸血症、枫糖尿病、异戊酸血症、线粒体病、高胰岛素血症、低血糖、低血钾等遗传代谢病的防治有研究。