




精选内容
-
医生:现在掌跖角化症有可行的治疗方案吗?

 银屑病百科交流荟
银屑病百科交流荟 2022年12月27日
2022年12月27日 80
80
 0
0
 0
0
-
医生您好,11岁的男童水源性掌跖角化症怎么治疗
 AD/湿疹名医科普馆
AD/湿疹名医科普馆 2022年12月15日20
0
0
2022年12月15日20
0
0
-
手湿疹/汗疱疹/掌趾角化症
1、病因先天性排汗异常+角化异常,有遗传因素父母也有,5-15岁重。环境因素玩摩擦、潮湿的东西容易起,如沙土、肥皂、泡水时间长、写字握笔紧等。气候湿热、情绪紧张、手足出汗多等。2、表现手指肚、手缝小水泡,自然吸收干涸后脱屑,露出红色、薄嫩新生皮肤,此时常感疼痛。可同时伴有手足多汗、干燥皲裂脱屑,皮肤肥厚3、外用药抗生素药物+中强效糖皮质激素。手足角质层厚,用药吸收少,只有面部1/30,用药注意厚涂,可保鲜膜封包半小时。
刘晓雁医生的科普号 2022年12月08日1790
0
2
2022年12月08日1790
0
2
-
残毁性掌跖角化症有药物可治疗啦
遗传性掌跖角化症是一种以手足皮肤过度增厚、角化为特征的遗传性疾病。其中,部分掌跖角化症可以出现手指、足趾屈曲、挛缩,甚至断裂,导致功能散失,又叫做残毁性掌跖角化症。Olmsted综合征是一种以多以残毁性掌跖角化及腔口周围角化,并伴有严重瘙痒及疼痛为特征的罕见皮肤遗传病,病人也可表现出广泛的脱发、关节挛缩、甲发育不良。Olmsted综合征并没有有效的治疗方法,临床常用的治疗角化性疾病的阿维A多数达不到满意效果,患者剧烈的疼痛及瘙痒往往无法完全缓解。2020年1月发表于JAMA Dermatology(美国医学会杂志,皮肤病学;国际知名医学杂志)的两篇文章介绍了Olmsted综合征的有效治疗新方法,为未来的治疗策略指出了新的方向。下面为病友们介绍最新关于Olmsted综合征治疗的学术进展。2012年,林志淼、杨勇课题组在国际上首次确定了瞬时受体电位通道V3基因(TRPV3)功能增强性突变引起了Olmsted综合征。后续研究者们证实TRPV3的激活导致EGFR(表皮生长因子受体)配体(即作用于表皮生长因子受体上的分子)的分解减少,从而使EGFR过度激活。因此,EGFR抑制剂理论上有可能缓解Olmsted综合征患者的症状。厄洛替尼是一种EGFR抑制剂,早在2004年,美国FDA就批准其用于非小细胞肺癌的靶向治疗,我国在2006年批准其临床应用,主要商品名是特罗凯。前述JAMA Dermatology两篇文章中的7例患者,都患有严重的Olmsted综合征,表现为严重的掌跖角化症及腔周角化,并伴有显著疼痛,身体活动严重受限,生长发育落后,且既往局部使用其他药物及口服阿维A效果不佳。在使用盐酸厄洛替尼治疗后,患者的症状(包括皮肤病变及瘙痒、疼痛)得到全部或基本全部缓解,患者可以站立或走路,心理状况及社会功能得到了明显改善(如下图所示)。 由于使用了抗肿瘤药物来治疗,不少患者会担心严重的不良反应。实际上,目前的靶向药物,和传统的细胞毒药物(也就是大家所了解的化疗药物)比较,不良反应是小很多的。该研究的患者年龄从1-17岁不等,在使用盐酸厄洛替尼的治疗过程中,在患者身上观察到的不良反应有——轻微的痤疮及脱发及指腹的轻微脱屑,这些患者的痤疮及脱发对局部用药的反应良好,指腹轻微脱屑的症状在调整药物剂量后消失。这些患者身上未观察到严重的药物不良反应。但其他类型EGFR抑制剂的有效性及治疗Olmsted综合征的用药剂量还需进一步研究。不过盐酸厄洛替尼的市场售价还是比较昂贵的。在上述的两篇文献中,药物的口服起始剂量为2mg/kg/天(在这7例患者中最高的起始剂量为100mg/天),如果服用这个药物,每个月的花费可能会高达万元以上。此外,治疗过程中要注意随访,医生会根据治疗效果和不良反应的发生随时调整用量。那么,什么样的掌跖角化症患者才适合上述的治疗方案呢,在确定治疗方案前又需要做什么呢?首先,患有掌跖角化症的患者需要通过与专业的皮肤科医生面诊并且进行基因检测以确定TRPV3是否为致病基因,再结合目前的症状、既往治疗方案及效果,才能确定是否合适使用盐酸厄洛替尼治疗。附:林志淼大夫出诊时间北京大学第一医院:周一上午、周三上午、周四下午优合诊所:周三夜间北京星宜诊所:周日上午本文作者:江星元博士,林志淼主任医师。参考文献:[1] Céline Greco, Stéphanie Leclerc-Mercier, Sarah Chaumon, et al. Use of Epidermal Growth Factor Receptor Inhibitor Erlotinib to Treat Palmoplantar Keratoderma in Patients with Olmsted Syndrome Caused by TRPV3 Mutations. JAMA Dermatol, 2020, Jan 2.[2] April Zhang, Sabine Duchatelet, Nikita Lakdawala, et al. Targeted Inhibition of the Epidermal Growth Factor Receptor and Mammalian Target of Rapamycin Signaling Pathways in Olmsted Syndrome. JAMA Dermatol, 2020, Jan 2.
林志淼医生的科普号 2020年01月25日10364
6
10
2020年01月25日10364
6
10
-
手指关节背部皮肤越来越厚、越来越硬,这是什么?
这其实就是——【指节垫】 手足指(趾)间关节的伸侧出现扁平或隆起的局限性角化损坏,表面光滑或粗糙不平。 形成的原因有很多,常见的有: 皮肤行为症(比如反复摩擦、搔抓、啃咬);还有一部分有家族史的与遗传有关。 如何处理呢:避免诱因,减少局部的反复刺激。可以外用维A酸乳膏或者肤疾宁贴膏改善增厚的皮肤,也可以在皮损地方注射类固醇激素混悬液,但以后也容易复发。 一般不建议手术切除,因为容易留下疤痕增生。
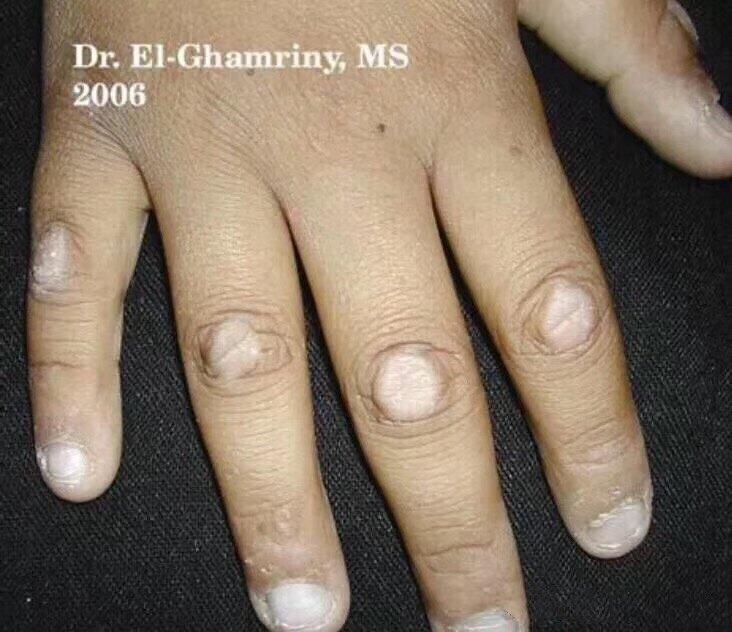 廖毅医生的科普号
廖毅医生的科普号 2019年10月17日9624
0
0
2019年10月17日9624
0
0
-
掌跖角化病的遗传
一、概述掌跖角化病(KPP)又称掌跖角皮症,是一组异质性显著的以掌跖皮肤增厚、角化过度为特征的一组慢性皮肤病。本病包括遗传性和获得性两种类型,遗传性疾病占大多数,多为常染色体显性遗传、故又称遗传性掌跖角化症。掌跖角化症属于一种常染色体显性遗传病,有明显的家族性,可代代相传或遗传数代终止,无阳性家族史的可追溯。可自幼发病,也可于儿童期或青春期发病,均有家族史。主要表现为掌跖皮肤角化的相关表现。此病因为遗传性而无法根治。本病亦可为症状性,仅为毛囊角化病及毛发红糠疹。一般预后较好。二、临床类型及表现按照皮肤损害形态的不同,分为弥漫性掌跖角化病(DPPK)、点状掌跖角化病(PPPK)、钱币状掌跖角化病及获得性掌跖角化病。1.弥漫性掌跖角化病:多于婴儿期发病,轻者仅有掌跖皮肤粗糙,严重时掌跖部出现弥漫性、边缘清楚的角化增厚性斑块,表面光滑,黄色,酷似胼胝,或呈疣状增厚,易发生皲裂,引起疼痛。可伴多汗、甲板增厚、浑浊等角化损害,终生不会自行消退。2.点状掌跖角化病:通常于20~30岁发病,皮损为掌跖部对称、散在分布的多呈圆形或椭圆形的角化性丘疹,高出皮肤,数目较多,一般直径仅为0.2~0.3cm,大者可达1cm。多呈灰黄色,质地坚硬,中心剥脱后形成火山形状的凹陷,可聚集成疣状角质损害。3.钱币状掌跖角化病:大多自幼(1~15岁)发病,皮损为过度角化性斑块或豆状角化过度,慢性进行性发展。皮损的发生部位及严重程度与机械摩擦及其强度有关。皮损为过度角化性斑块或豆状角化过度,慢性进行性发展,多见于掌跖侧缘。甲下、甲周也可见角化过度。跖部较掌部多见且严重。自觉疼痛,严重者影响行走。4.获得性掌跖角化病:四肢的淋巴水肿性角化病,掌跖部出现弥漫角化过度,其表现有疣状突起及皲裂。主要见于大鱼际和小鱼际。三、病因1.遗传性因素 大部分为常染色体显性遗传,少数为常染色体隐性遗传。2.获得性因素 银屑病、湿疹、手足癣、扁平苔藓等皮肤病,内脏器官癌及应用砷剂均可出现掌跖角化病。病因根据疾病类型而异,如绝经期掌跖角化病及掌纹点状掌跖角化病等属于原因不明的掌跖角化病;砷掌跖角化病与接触砷剂有关;而银屑病、慢性湿疹、毛发红糠疹等疾病也可出现掌跖角化的表现。四、遗传学诊断与咨询(一)遗传学诊断目前发现弥漫性掌跖角化病的主要致病基因为KRT1及KRT9;点状掌跖角化病的致病基因为AAGAB及COL14A1。检查患者血液或皮肤组织样本中的基因,即可明确诊断。(二)遗传咨询1.按照常染色体显性遗传方式咨询。2.患者双亲风险评估 患者双亲有一方是正常人,另一方是患者,该患者一般是杂合子,也许是纯合子患者。3.患者同胞风险评估 如果患者的双亲中的患者为杂合子,其同胞中有1/2的为患者,与性别无关;如果患者的双亲中的患者为纯合子,其同胞中均为患者。4.患者后代风险评估 (1)患者与正常人结婚,其后代中有1/2的为患者,与性别无关。 (2)如果患者与一杂合子患者结婚,其后代中有3/4的为为患者,与性别无关。(3)如果患者与纯合子患者结婚,其后代中100% 的为该病患者,与性别无关。
窦肇华医生的科普号 2019年09月24日5682
0
3
2019年09月24日5682
0
3
-
知“足”常乐-----解析那些令人困扰的足部皮肤病
---------谈谈那些长在脚上的让你不快乐的皮肤病人都说随遇而安,知足常乐,可是一旦足部皮肤病不期而遇的来到,你又不了解它,就乐不起来了。足部的面积只占全身皮肤总面积的7%左右,但能生的皮肤病却不少,让人困扰不堪,下面Dr.liang就来跟大家谈谈足部皮肤病:1、足癣足癣俗称“脚气”,是最常见的皮肤病之一,但也是最让人困扰的一种足部皮肤病,它是由真菌感染引起的,潮湿、闷热的时候容易发,主要表现为脚痒、脱皮、水疱等,临床上常分为角化过度型,浸渍糜烂型,水疱型,丘疹鳞屑型等。抗真菌治疗有效。平时要勤换鞋袜,勤洗脚,保持鞋袜透气,才能防止复发。2、湿 疹湿疹是过敏引起的皮肤病,全身都会发,脚上当然不例外,常与食物、灰尘、鞋子颜料、胶水等过敏刺激有关,分急性和慢性,急性常有红斑、渗液,慢性主要是增厚及脱屑。湿疹常常双足对称发生,境界不清,真菌镜检阴性。口服抗过敏药及外用卤米松等有效,有时与足癣不易区分,有时湿疹可合并足癣,自己切勿乱用药,应至医院就诊治疗。3、汗疱疹春夏季易发,与环境过敏及精神因素等有关,表现为密集深在性小水疱,瘙痒剧烈。调节情绪、避免刺激、抗过敏治疗及外用激素药膏有效。4、跖疣跖(读zhi,第二声)疣是发生在足底部的寻常疣,由人类乳头瘤病毒(HPV病毒)感染引起,老百姓俗称“刺瘊”“瘊子”“千日疮”。跖疣具有传染性,主要是自身传染,长了一个之后很容易变成三五个,甚至更多。冷冻治疗效果较好,不留疤痕,也可以用激光、手术、干扰素、药物腐蚀等。5、鸡眼鸡眼是足部皮肤长期受压和摩擦引起的圆锥状角质增生,行走较多、脚骨异常的人较易发生,摩擦和压迫是主要诱因。很多人对鸡眼和跖疣傻傻分不清,其实很容易分别:鸡眼是圆形蜡黄色的丘疹,中央可见硬的角质芯;跖疣表面粗糙,常有散在黑点。鸡眼可以用手术剜除、冷冻或鸡眼膏治疗。穿软底鞋子,避免挤压是关键。6、胼胝(pián zhī)俗称老茧,也是足底皮肤受压或摩擦引起,成片的角质层增厚,不像鸡眼那么疼痛,胼胝通常没有症状,可以用热水泡脚后自己修剪去除。7、丹毒丹毒是一种累及真皮浅层淋巴管的感染,主要致病菌为A组β溶血性链球菌,细菌常从足癣所致的微小皮肤伤口入侵,继而发生感染。常伴有畏寒、寒战、发热,不仅足背累及,且很容易蔓延至小腿伸侧,表现为红肿热痛。青霉素类药物治疗有效,疗程较长,需10~14d,治疗期间尽量卧床休息,抬高患肢,有足癣的应抗真菌治疗,防止丹毒复发。8、癣菌疹癣菌疹是皮肤对足癣等感染时释放的真菌抗原表现出来的急性过敏反应,瘙痒剧烈,短时间内会蔓延到远处部位,应该立即去医院就诊,抗过敏治疗为主,有时需使用小量激素才压得住病情。9、丘疹性荨麻疹与昆虫叮咬有关,夏秋季穿着衣物较少时容易发,足部离地面近,尤其是调皮的小朋友,更容易被昆虫叮咬,表现为红斑、丘疹,伴瘙痒,有时候有水疱,需要与水痘鉴别。口服抗组胺药及外用炉甘石洗剂,避免搔抓。10、手足口病此病早已名声在外,大多由柯萨奇病毒A16型感染所致,小儿多见,大人偶发也会发。主要症状如其名:手、足、口部位出现小水疱,此外臀部也是水疱好发部位,但是有时候症状不典型,只有一处或两处起了水疱。其实手足口病大部分病情都很轻,对症治疗就能恢复。少数由肠道病毒71型感染所致的手足口病较重,会出现病毒性脑炎、心肌炎等严重并发症。因此,为了万无一失,得了手足口病,还是应该去医院正规治疗。11、二期梅毒疹此图很经典,无须解释,正规有经验的皮肤科大夫一看即知。洁身自好的正常人都不会得的;假如你近期有过无保护高危性接触,而足底和手掌都长了椭圆形古铜色伴脱屑的斑,那还是赶紧去医院皮肤科做梅毒化验吧!12、冻疮冬天时候,天气寒冷皮肤血管收缩,缺血缺氧,使局部皮肤产生水肿性紫红斑、水疱,有时还会糜烂。足部远离心脏,和手指、耳朵是难兄难弟,都是冻疮好发部位。最主要是注意保暖,增强体质。病情严重的需要口服扩血管药物治疗。13、甲癣俗称“灰指甲”,是真菌感染趾甲引起的,常和足癣并发,有传染性,治疗起来却比足癣困难得多,部分人外用冰醋酸有效,大部分人需要口服伊曲康唑等抗真菌药治疗,此类药很多人闻之色变,其实大部分没有肝脏基础疾病的人吃了还是很安全的,而且效果可靠。14、甲沟炎由于鞋子压迫或修剪指甲不当等引起的甲沟皮肤软组织的细菌性感染,红肿疼痛,还有化脓,需要清创、换药及抗生素治疗,个中苦楚只有得过的人才能体会。还有一种念珠菌性甲沟炎,也红也肿,但一般很少流脓,也不像细菌性甲沟炎那样疼痛,常见于有咬手指癖的儿童和常接触脏水的人,需要抗真菌治疗。15、其他甲病:其实小小的趾甲能得的病还真多,除了最常见的甲癣,还有甲母痣(甲黑线)、甲纵嵴、甲横嵴、先天性厚甲症、嵌甲、甲营养不良、甲下血管球瘤、甲扁平苔藓、甲下外生骨疣等等,所以甲病还是要到正规医院就诊,不能全当灰指甲一治了之。16、环状肉芽肿一种少见的比较奇特的皮肤病,足背好发,表现为小丘疹或结节组成的环状隆起,病因不明,可能与外伤、昆虫叮咬、日光照射、药物、病毒感染等有关。17、慢性溃疡慢性溃疡是常见病多发病,足部也是好发部位,与感染、糖尿病、营养状况、血管情况等都有关,皮肤破溃长时间不愈合,严重影响日常工作生活,久之还可能继发皮肤癌,不是随便擦擦某秘方膏药就能治好的,需要医院寻找病因,对症治疗。18、掌跖脓疱病如果你足底手掌经常反复发作小脓疱,然后有红斑、脱皮,当做湿疹或者脚气怎么也治不好,就要当心这个病了。掌跖脓疱病现在有人认为是银屑病(俗称牛皮癣)的脓疱型的局限亚型,用维A酸联合紫外线光疗效果显著。19、窝状角质松解症一种看起来比较令人不适的脚脱皮,是棒状杆菌属引起的跖部角质层剥蚀的一种皮肤病,热带亚热带地区接触泥土时被感染,红霉素或四环素治疗有效。20、掌跖角化症为限局性足底、手掌角化、肥厚,常自幼发病,是一种常染色体显性遗传病,只能用保湿、去角质等对症治疗改善症状。21、 更年期角化症本病仅发生于女性,于绝经前期或绝经期发病,足跟、跖缘等受压及摩擦部位干燥、角化、粗糙、肥厚,外用尿素乳膏封包治疗有效。22、交界痣色素痣有很多种,长在脚底的主要是交界痣,呈平坦的褐黑色小斑片,很多人看了《非诚勿扰2》里面李香山脚背上隆起的黑素瘤之后到医院要求祛痣。其实电影里面黑素瘤的部位和形态都是很业余的,那是足背的皮内痣,恶变几率极小的,足底的交界痣虽然恶变几率比皮内痣高,但也还是很低很低的。大家到医院来咨询、定期观察是对的,过度紧张就没必要了。23、恶性黑素瘤真正的黑素瘤是一种恶性程度非常高的肿瘤,在我国恶黑原发部位不少都在足跖,那么什么样的痣可能有恶变的前兆呢?一般有如下情况应立即去医院就诊:短期内痣颜色加深、变黑、增生、隆起、破溃、出血、出现卫星灶等都是不好的预兆。现在有专家总结了需高度怀疑恶黑的痣的ABCDE特征,便于自己对照观察:A(Asymmetry):不对称, B(Border irregularity):边界不规则,C(Color):颜色不均一,甚至可以出现蓝、灰、白、红色。D(Diameter):黑色素瘤的直径常常大于6mm,E(Elevation):皮损隆起、进展。24、色素沉着-息肉综合征又称Peutz-jeghers综合征,为常染色体显性遗传疾病,皮疹表现为口唇、足跖等处的褐黑色斑点,伴小肠腺样错构瘤,肠道息肉少数可以恶变。所以若足趾、口唇有色素斑,又经常有腹痛、黑便的,应去消化内科做肠镜检查。25、其他皮肤病:遗传性对称性色素异常症、放线菌性足菌肿、皮肤结核、小儿丘疹性肢端皮炎、阿洪病、过敏性紫癜、网状青斑、雷诺氏病、闭塞性动脉硬化症、闭塞性血栓性脉管炎、化脓性肉芽肿、多形红斑、白癜风、带状疱疹等。皮肤病共有1800余种,足部的皮肤病也有上百种,全部写完的话可以成一本书了.诊断足部的皮肤病有时并不容易,需要结合患者足部皮疹的形态特点,其他部位皮疹的形态,患者的职业,生活习惯等,必要时还需化验检查,病理活检,才能确诊。文章有点长,大家没病的可以科普下,有足病困扰的可以对号入座,了解个大概,如果对不上号或自己看晕了的,还是尽快来医院找专业皮肤科医生帮忙吧。医者仁心,知“足”常乐。梁宁 主治医师原创文章,转载请注明出处。(部分图片来自网络)本文系梁宁医生授权好大夫在线(www.haodf.com)发布,未经授权请勿转载。
梁宁医生的科普号 2016年12月16日93345
18
52
2016年12月16日93345
18
52
-
中国人最常见的遗传性掌跖角化症--长岛型掌跖角化症
长岛型掌跖角化症,又称Nagashima掌跖角化症,是一种日本人首先命名并发现致病基因(SERPINB7)的一中遗传性掌跖角化病,主要表现为双手、双足的皮肤红斑、增厚,伴有遇水后掌跖角质发白、肿胀,及手足多汗。足部多汗通常会继发真菌感染,部分患者还伴有肘部及膝盖部位的皮肤红斑、角化。本病为SERPINB7基因突变所导致的常染色体隐性遗传性疾病,由于中国人和日本人中携带SERPINB7致病基因的比例较高(多达3%),因此这个疾病在中国和日本发生率很高,按照我们的论文研究报道,本病可能是我国国人最常见的遗传性掌跖角化症。本文发表在国际皮肤科专业顶级杂志,研究性皮肤病杂志(Journal of Investigative Dermatology),为我研究组在国内首先发现的7例遗传性长岛型掌跖角化症患者的致病基因,以及基因突变位点的来源。对于长岛型掌跖角化症的认识,可以解开许多患者心中最关心的迷:到底我们的掌跖角化症会不会遗传?孩子以后会不会出现牙齿异常?指甲异常?甚至出现手足指趾的断裂挛缩?本文可以给大家部分答案。若仍有其他问题,可以与我(北京大学第一医院皮肤科林志淼医生)进行交流。
林志淼医生的科普号2015年02月04日30026
14
3
-
掌跖角化病如何治疗?
患者:病情描述(发病时间、主要症状、就诊医院等): 我从刚出生到现在已经29年了。手脚掌发红发硬有时候还脱皮、皮肤纹理特别深看起来手掌脚掌皮肤比较粗糙老化。尤其是脚掌。皮肤特别硬和厚。都有些发黄了。每次洗完脚了死皮特别厚。用刀子轻轻一刮就能挂掉。过2天又长的很厚。还脱皮从来不敢和别人握手感到很自卑。。。。夏天也不敢穿凉鞋。。。。 小时候父母带我去过北京几个比较大的医院。医生都说是遗传先天性的不能治好。可那是很多年前的事情了。不知道现在有没有什么好的办法治疗或缓解。至少让皮肤和正常人的皮肤一样。。谢谢 希望能帮到我。。让我的手掌和脚掌皮肤能和正常人的一样。。。解放军总医院第一附属医院皮肤科邹先彪:你好!掌跖角化病(PPKs)是一组包括不同病种的疾病,这类疾病以手掌和足底皮肤的异常增厚为特征。传统上我们把角化病分类为遗传性和获得性,它们之间通过以下几方面相区别,包括遗传模式、逾越现象的存在(即超越手掌和/或足底皮肤外角化过度的连续延伸)、其它并存疾病以及表皮累积范围,即弥漫性、局限性、斑点状。获得性PPKs一般分类为:绝经期皮肤角化病、药物性PPKs、营养不良性PPKs、化学性PPKs、系统性PPKs、恶性肿瘤性PPKs、皮肤病性PPKs、传染性PPKs及原发性PPKs。 需要面诊寻找可能的潜在病因,如果并未发现明确的原因,那么保守疗法包括局部使用角质层分离剂(尿素、水杨酸、乳酸)、重复的物理清创术、局部维甲酸治疗、局部补骨脂素+UVA疗法及局部激素治疗。对于个别顽固病例,依曲替酯和阿维A做为一种替代疗法也可取得良好疗效。
邹先彪医生的科普号 2010年06月23日14182
27
0
2010年06月23日14182
27
0
掌跖角化病相关科普号
窦肇华医生的科普号
窦肇华 主任医师
北京家恩德运医院
遗传咨询科
6289粉丝280.6万阅读

汪旸医生的科普号
汪旸 主任医师
北京大学第一医院
皮肤性病科
6718粉丝107.6万阅读

秦明珠医生的科普号
秦明珠 副主任医师
淮北市人民医院
皮肤性病科
3550粉丝82.8万阅读
-
 推荐热度5.0周俊 主治医师西安交大二附院 皮肤科
推荐热度5.0周俊 主治医师西安交大二附院 皮肤科激光美容 97票
鲜红斑痣 67票
雀斑 45票
擅长:1. 鲜红斑痣的光动力治疗 2. 皮肤激光与光子治疗美容(包括祛斑、毛细血管扩张、嫩肤、瘢痕、脱毛、除皱等) 3.肉毒素注射 4.皮肤损容性皮肤病变(痤疮、汗管瘤、睑黄瘤、瘢痕等)的治疗。 5.光老化、皱纹、面部皮肤年轻化方案设计 6.腋臭的治疗 7.皮肤科常见病与多发病的诊治。 -

 推荐热度4.9马东来 主任医师北京协和医院 皮肤科
推荐热度4.9马东来 主任医师北京协和医院 皮肤科白癜风 491票
皮肤病 32票
湿疹 10票
擅长:①色素性皮肤病(尤其是白癜风和外阴白斑);②遗传性皮肤病的临床和研究工作;③各种疑难、少见和重症皮肤病的诊断和治疗;④感染性皮肤病;⑤皮肤组织病理;6.卟啉病的诊断和治疗。 -
 推荐热度4.7李明 主任医师复旦大学附属儿科医院 皮肤科
推荐热度4.7李明 主任医师复旦大学附属儿科医院 皮肤科特应性皮炎 20票
皮肤病 9票
脱发 4票
擅长:儿童湿疹,咖啡斑、鱼鳞病、掌跖角化病、少毛症等遗传性皮肤病以及真菌、细菌、病毒等感染性皮肤病的诊治。