提 要 神经纤维瘤病是一种家族性遗传性疾病,易造成患者不同程度残疾。本文针对该病的分类、临床表现、诊断及治疗进行综述,以期对指导临床有一定帮助。
关键词 神经纤维瘤病 分类 诊断 治疗
神经纤维瘤病(neurofibromatosis)又称多发性神经纤维瘤。由法国病理学家Von Recklinghausen 1882年首先报告本病,故通常称为Von Recklinghausen病。是一种家族性遗传性疾病,有染色体显性遗传倾向,发病率约占出生儿的三千分之一。其基本病变为神经组织发育不良,全身各个器官和系统都可受累[1]。
1 分类
通常将神经纤维瘤病分两型,即神经纤维瘤病Ⅰ型和神经纤维瘤病Ⅱ型。神经纤维瘤病Ⅰ型(NF1),即周围性神经纤维瘤病,又称Von Recklinghansen 病,新生儿或儿童早期表现明显,表现为多发性神经纤维瘤病,皮肤上有片状棕褐色的色素沉着斑或叫咖啡斑(cafe-au-lait spot),眼上可出现Lisch结节,部分患者中枢神经系统受累,可出现语言障碍,智力减退,巨大头(macrocephaly),骨骼异常,恶性变等。神经纤维瘤病Ⅱ型(NF2),又叫中心性神经纤维瘤病或双侧听神经纤维瘤病,常在10岁以后才出现明显的表现,发生双侧听神经的肿瘤(双侧听神经瘤)。
NF1和NF2由两种完全不同的基因所引起,NF1基因位于第17染色体上,已经在1990年被克隆并编码为神经纤维瘤蛋白(neurofibromin),神经纤维瘤蛋白的真正作用目前还不了解,但它包含单个显性鸟苷三磷酸(GTP)酶激活功能。NF2基因位于第22染色体上,于1993年被克隆出来,命名为局部蛋白(merlin),其真正的作用目前仍不清楚[2-6]。
2 临床表现与诊断
2.1 神经纤维瘤病Ⅰ型
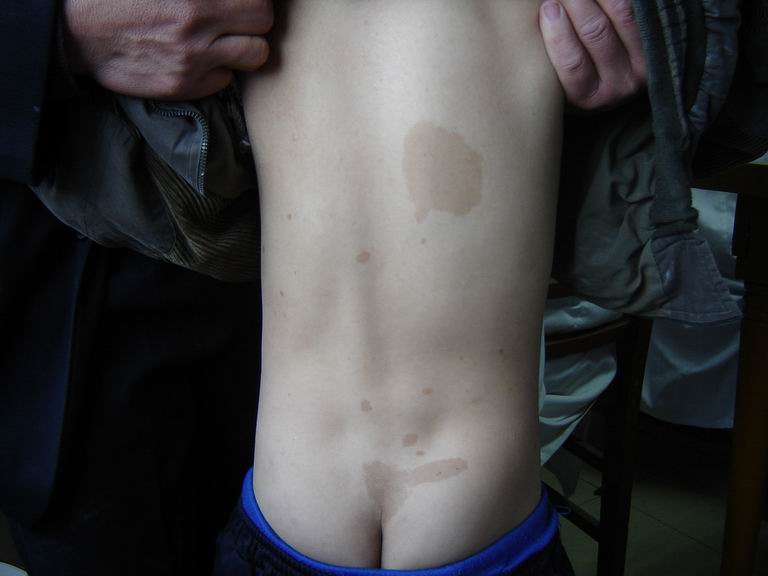
2.1.1 皮肤褐色斑或咖啡斑:正常人10-20%也可出现皮肤褐色斑,而95%神经纤维瘤病患者可出现此斑。78%NF1患者可出现6个以上的咖啡斑,直径大于5cm。本病DOPA阳性的出现率高于周围皮肤,而正常人所出现的褐色斑、以及其他疾病(Mclune-Albright综合症)所出现的咖啡斑每平方厘米DOPA阳性黑色素细胞较少。另外,本病所出现的斑点边缘光滑,呈淡褐色,而Mclune-Albrighy综合症斑点边缘呈暗褐色,边缘不规则。
2.1.2 Crowe`s征:20-50%患者腋部或腹股沟出现雀斑(freckle),直径1-4mm,多数NF1患者长有3个以上这种雀斑[7]。其他皮肤皱折部位也可出现,如颌下、女性的乳房下。有人报告6岁患者81%有腋部或腹股沟雀斑[8],它是仅次于咖啡斑的诊断儿童NF1的表现,所以它对儿童初期和青少年前期的NF1诊断非常重要。
2.1.3 兰红色斑点和假性萎缩性斑点(Blue-red macules 和 pseudoatrophic macules):一般出现在青春期,斑点轻微高出皮肤,内含小血管及扩张的毛细血管。
2.1.4 神经纤维瘤(neurofibromas):神经纤维瘤病为错构性的具有多细胞起源,多有雪旺细胞组成,但也含有成纤维细胞、巨细饱和巨噬细胞,多在青春期出现,可为数个至数百个,大小各异,数目与大小随年龄增长而增大,女性妊娠期明显增大、增多。另外,女性患者在青春期后常出现乳晕的神经纤维瘤。
2.1.5 Lisch结节:是NF1常见的表现,这些虹膜错构瘤呈穹隆形,高出皮肤的、无血供的黑素细胞的结节,边缘光滑、质软,颜色从浅褐色到深褐色,直径通常小于2cm,在双目生物显微镜下最易看清。
2.1.6 癫痫发作和智力障碍:NF1患者通常头颅较正常人大[9、10]。神经病变占患者的50%,包括癫痫发作、智力障碍和肿瘤。癫痫发作占8-13%,常常与颅内肿瘤有关。智力障碍(包括延迟发育、机能亢进、学习障碍和语言障碍)占10-20%,其中仅有2-5%的患者有轻度精神发育迟缓,30-4-0%将有语言残疾。
2.1.7 视力和听力影响:儿童早期出现眼睑下垂是眼睑神经纤维瘤或丛状神经瘤的预兆。视神经胶质瘤、神经纤维瘤、雪旺氏纤维瘤和视神经鞘膜瘤可侵犯眼球和周围软组织。青光眼是少见的并发症,视神经胶质瘤在生后10年内易于出现,表现肉瘤样退变、神经周围生长方式。
2.1.8 颅内和脊髓肿瘤:与神经纤维瘤病有关的颅内肿瘤有视神经胶质瘤、星形细胞瘤、神经鞘瘤、听神经瘤、神经纤维瘤病和脑脊膜瘤病。常见的脊髓肿瘤为脊膜瘤或神经纤维瘤。
2.1.9 恶形肿瘤:神经纤维瘤病的恶变率为1-15%,常为肉瘤变,即形成神经纤维肉瘤,其他肿瘤包括白血病、恶性神经鞘瘤、Wilms肿瘤和嗜络细胞瘤[11]。
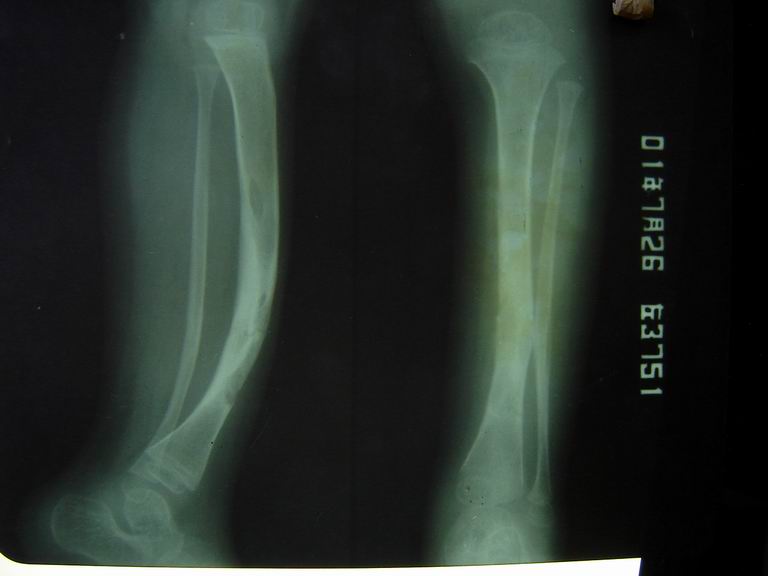
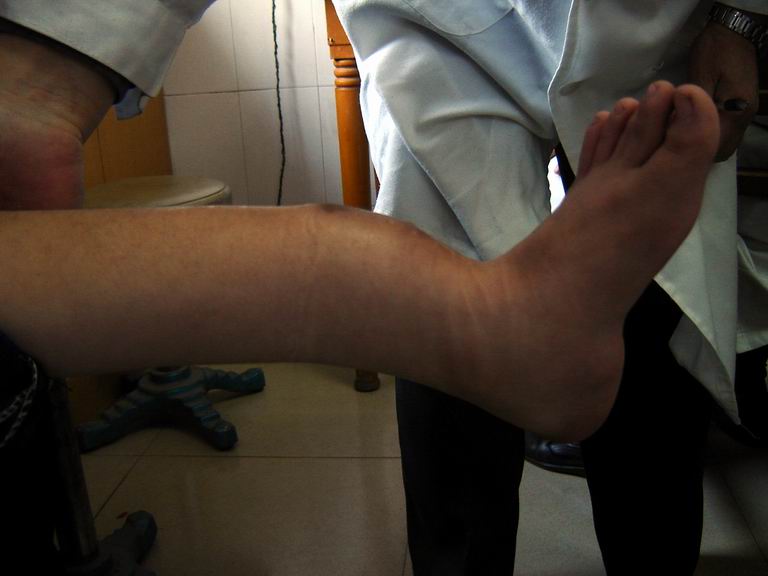
2.1.10 骨缺陷和先天性脱位:神经纤维瘤病患者中一半可能存在骨缺陷,包括脊柱后突、侧突等畸形(约10%)、假关节形成和长骨骨皮质发育不良或变薄,这种改变均与骨内或临近神经纤维瘤的破坏有关。
2.1.11 口腔病变:约72%患者存在口腔病变,常见的有口腔软组织神经纤维瘤(27%)、增大的蘑菇状乳头(32%)、下颌骨损害(18%)、牙槽腔增宽(27%)和下颌骨孔增大(27%)。
2.1.12 内分泌异常:性早熟或性晚熟(以前者多见),月经异常,男子女性型乳房,肢端肥大症,甲状腺机能亢进或低下,甲状旁腺机能亢进,甲状腺髓内肉瘤、不生育、Addison病和低血糖。
2.1.13 胃肠道受侵:10%患者有如下并发症发生,小肠神经纤维瘤、表面粘膜出血、消化不良、呕血、黑便、肠梗阻和肠穿孔等。
2.1.14 高血压:成人神经纤维瘤病患者发生高血压者并不少见,其原因可能是肾血管性、也可能是嗜络细胞瘤所致或两者兼有。18岁以下以肾血管性多见,为嗜络细胞瘤所致高血压的7倍,NF患者中嗜络细胞瘤发生比例是1:223,而嗜络细胞瘤患者中NF的发生比例在1:5到1:20之间。
2.1.15 血管异常:几乎所有NF患者有血管损害,最常受累的是腹主动脉、肾动脉、颈内动脉和椎动脉。儿童最常出现的是先天性肾血管异常。肾动脉狭窄源于血管中层的纤维肌肉发育不良,最先发病的部位在肾动脉与腹主动脉的起始处,然后向肾内血管分支蔓延。
神经纤维瘤病Ⅰ型的诊断标准:该标准是由1987年美国国家健康研究所(NIH)制定的,若具备下列内容之2条或2条以上,即可诊断为NF1:⑴ 6个或6个以上咖啡斑,青春期之前患者,直径大于5mm,青春期之后直径大于15mm;⑵ 2个以上任何类型的神经纤维瘤或1个丛状神经纤维瘤;⑶ 腋部或腹股沟部多个雀斑(Crown征);⑷ 视神经肿瘤;⑸ 2个以上Lisch结节(虹膜错构瘤);⑹ 不同与其他疾病的骨损害,如长骨骨皮质蝶翅状发育不良或变薄,伴有或不伴有假关节存在;⑺ 1个一度亲属(父母、兄弟姐妹或子女)按前述标准患有NF1。
2.2 神经纤维瘤病Ⅱ型的诊断标准 NIH会议对NF2的诊断标准有:⑴CT或MRI发现双侧第8脑神经肿物,与听神经瘤表现一致;或⑵一度亲属中有1位患NF2和下列两种情况之一, a单侧第8脑神经肿物;b具有下列之2种病变:神经纤维瘤、脑脊膜瘤、胶质瘤、神经鞘瘤或后方被膜下晶状体混浊[12]。新的NF2诊断标准与前述标准稍有不同:⑴MRI发现双侧前庭神经鞘瘤;⑵父母、兄弟姐妹或子女之一患NF2,加上(a)30岁之前诊断为单侧听神经神经鞘瘤或(b)下列病变之2:脑脊膜病、胶质瘤、神经鞘瘤,青少年后方被膜下晶状体浑浊。在临床上,这些诊断标准太教条,以致于可能将这些NF2患者排除在外[13、14]。NF2患者可出现视网膜错构瘤或皮质楔形浑浊,视网膜错构瘤可出现于较年轻的患者,对早期诊断有帮助,NF2中达9%-22%[15-17]。
2.3 NF1和NF2基因诊断的应用
大多数NF1的诊断单纯依靠临床表现可以确定。50%的NF1是因新的基因突变所致,传统的识别NF1突变基因受到限制,这是由于NF1基因较大且突变是千变万化的,未发现突变集中在基因的任何一个区域。NF1基因跨越基因DNA350Kilobase以上,由60个 外星子(exon)组成,所以在DNA中检查突变位点需要大量的技术工作,即费时,价格又非常昂贵。
目前基因突变检测已经出现,用蛋白切断试验(protein truncation test,PTT)将来自白细胞的RNA翻录并转变重叠NF1互补的DNA片段,以作为模板用于分析神经纤维瘤蛋白片段。每一个神经纤维瘤蛋白片段根据大小被分离出来,对这些片段进行识别分析以确定NF1突变与否。不幸的是NF1因家谱不同表现变化很大,而这些检验只能告诉我们该个体是否患有NF1的危险,而不能了解其严重程度,这限制了该技术用于出生前家族性普查的应用,将来的研究可能会促进DNA或生物化学诊断技术的提高。
NF2的基因诊断同样面对这些困难或问题。一旦基因突变被认可,对该家庭进行特殊检查非常必要。但同样需耗费大量的时间和费用。目前研究集中在判定NF2基因突变与临床严重程度的关系上[18]。不管怎样,神经纤维瘤病基因的识别为我们从分子基础上了解该病开阔了视野,将来的研究将针对用基因疗法治疗该类疾病。
3 观察与治疗
3.1 神经纤维瘤病Ⅰ型的观察、治疗
3.1.1 婴幼儿期:咖啡斑可在出生时出现,也可在生后1-2年内出现,丛状神经纤维瘤可能是先天性的,但常常在出生后第一年发作,一般无须治疗。胫骨发育不良可在出生即出现下肢向前外侧弯曲,应给予保护、固定,以防止骨折的发生。
3.1.2 学龄前:3-5岁之间开始出现雀斑,也可发生神经胶质瘤,所以10岁以内应经常找有经验的眼科医生检查,任何视神经异常即应行颅内MRI检查。视神经肿瘤是NF1常见的表现,尽管其很少发展到视力丧失。若视神经肿瘤进展迅速,应行外科治疗[19]。但外科治疗这些肿瘤的作用是有限的,有时可联合应用化疗、放疗等[20],大剂量放疗或化疗后有发生恶性变的倾向。
学习障碍也是NF1最常见的表现之一,可占NF1的40-60%[21]。行为能力、运动协调能力均低于正常儿童。应定期检查评估患儿的血压,因为高血压也是NF1的又一表现,多源于肾动脉狭窄。丛状神经纤维瘤在儿童早期即可出现,有时呈自限性,该瘤难以完全切除,易于复发。部分NF1患儿可发生偏头痛,可伴恶心、腹痛,但需排除其他原因所致的头痛、腹痛,可应用药物预防和治疗。
3.1.3 儿童后期、青少年、青春期之前,皮肤神经纤维瘤表现一般比较典型,脊柱侧凸可在儿童后期或青少年时期出现,需要进行短节段脊柱融合。一部分患者出现先天性胫骨前外侧弓状畸形和先天性胫骨假关节,前者易发生骨折而继发假关节形成。如果有胫骨前外侧弓状畸形而未发生骨折,应该先应用支具保护,一旦发生骨折需要长时间石膏固定,也可行手术切开复位内固定加植骨术,但3岁以下患儿植骨术也难以愈合,有些行多次植骨方取得成功[1]。先天性胫骨假关节治疗更困难,有用电刺激、带血管腓骨移植等方法,但仍有一些患者需行膝下截肢术[1]。
3.1.4 成年人:皮肤神经纤维瘤的数量和部位难以预测,一些特殊部位的病变可被切除。有人应用二氧化碳激光治疗皮肤神经纤维瘤和咖啡牛奶斑。有高血压者应进行治疗。NF1发生恶性肿瘤的危险性有5%。恶性周围神经鞘瘤有时源于丛状神经纤维瘤,所以丛状神经纤维瘤若生长迅速或出现疼痛,应尽快手术治疗,并检查有无转移发生。
3.2 神经纤维瘤病Ⅱ型的观察、治疗
所有被诊断为NF2的患者均应定期进行头颅及脊髓CT或MRI检查,以了解内耳、脊髓、马尾有无肿瘤。对那些一度亲属中患NF2者亦应定期检查。视、听功能的定期评估也很重要,眼科医生、耳科医生、神经外科医生、骨科医生和基因专家将共同努力对这些患者及其亲属进行随访、观察、治疗。对听神经鞘瘤进行早期手术或是等完全出现耳聋后手术是有争议的也是十分关键的[22]。小的听神经瘤(〈1.5cm〉可被完全切除,并保留听神经和面神经功能[22]。较大的肿瘤通常采取切除主体或行神经减压术。其他的颅内或脊髓肿瘤,如脑脊膜瘤、颅神经神经鞘瘤、室管膜瘤也须进行随访观察,但这些肿瘤一般生长比较缓慢。
立体定位放射外科(stereotactic radiosurgery)即伽玛刀(gama knife)可用于治疗某些听神经鞘瘤[23]。但对NF2的其他肿瘤应慎重应用,因为对那些具有无活性肿瘤抑制基因的患者放射可使肿瘤诱发、加速或转变。
本文是陈清汉版权所有,未经授权请勿转载。 本文仅供健康科普使用,不能做为诊断、治疗的依据,请谨慎参阅










