医学科普
发表者:王忠诚 人已读
无肛症也叫肛门闭锁症,或肛门直肠异常。无肛症是新生儿比较常见的先天性疾病,约3000个新生儿有一例。在胚胎时期泌尿生殖及直肠系统都由共泄腔(cloaca)发展形成。于胚胎第五周起共泄腔中隔(urorectal septum)逐渐形成,而在第九周时,泌尿生殖系和直肠即完全分开。如果在上述演变过程中发生进展中止,则肛门直肠无法下降到正常的位置,造成直肠盲端或者肛门盲端合并有泌尿生殖道或肠道廔管。
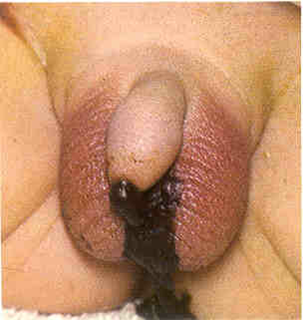
无肛:直肠与尿道相通
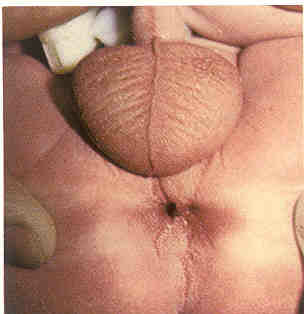
无肛门伴直肠会阴瘘
分类
临床上根据直肠盲端位于耻骨直肠肌(puborectalis)之上或下而分为高位与低位无肛。在女婴常见有瘘管通到会阴或阴道,男婴则常有瘘管与泌尿道相通。
1984年国际分类法(Wingspread)
高位型,直肠盲端位于提肛肌以上;
中间位型,直肠盲端为耻骨直肠肌包绕;
低位型,直肠盲端通过耻骨直肠肌。
检查
一、检查病人的肛门会阴处可看出没有肛门开口或者有小于正常的开口位于会阴、阴道或前庭(vestibulum)。没有廔管和体外相通的病人则会有较明显肠阻塞的现象。
二、部份没有瘘管的病人需于出生后24小时照"倒立姿势"的腹部X光片。如见充气之直肠盲端位于耻骨尾骨连线(相当于耻骨直肠肌之位置)之远端,属于低位型肛门闭锁症。如充气之直肠盲端位于耻骨尾骨连线之近端则为高位型肛门闭锁。
三、以逆行性膀胱尿道造影术或以显影剂注入直肠盲端,可以检查是否有瘘管和泌尿系统相通。此外,如于尿液中或于阴道中发现有胎便亦表示有瘘管存在。
治疗
治疗之要点在于重建正常的肛门开口,并保存其大便自制力(continence)。
一、低位型肛门闭锁症合并有异常之开口于会阴、前庭者,如有开口狭窄之情形,于出生后可予先扩张,则病人可以正常的排泄,等数个月后再行肛门成形术( anoplasty) 。
二、低位型肛门闭锁症无开口于体外,且盲端距正常肛门位置在两公分以内者。以肛门成形术,直接由会阴处手术将直肠分离,拉出于正常肛门位置。
三、高位型或低位型但盲端距正常开口在两公分以上者,则先以下结肠造口术解除大肠阻塞的现象。约一岁左右再行确定性手术。
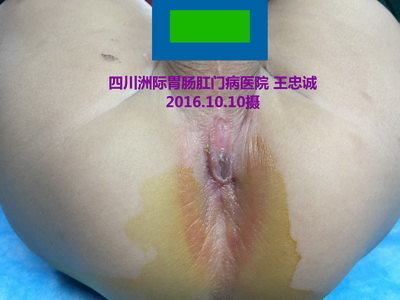
术前:小儿无肛伴直肠会阴瘘
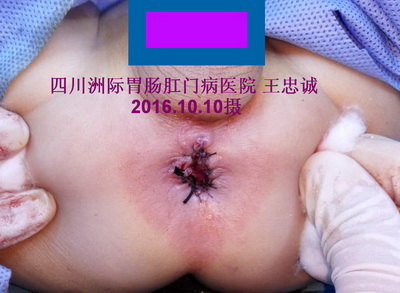
术后:肛门整形良好,术后第一天解出食指粗的成形大便。术后1周出院,1月后随访肛门正常,大便正常。
预后
低位型肛门闭锁症的病人,于手术后都能获得良好的大便自制力。高位型的病人,由于耻骨直肠肌本身的发育较差,因此手术后仍有部份的病人有大便之问题。定期随访检查肛门大小,以避免发生狭窄。
本文为转载文章,如有侵权请联系作者删除。本文仅供健康科普使用,不能做为诊断、治疗的依据,请谨慎参阅
发表于:2016-11-05










