



肿瘤患者代谢改变及其机制
摘要:恶性肿瘤发生中重度营养不良的发生率非常高,中国肿瘤患者的中度和重度营养不良发生率为58%,营养不良明显影响肿瘤患者的临床结局,近25%的肿瘤患者直接死于营养不良。许多营养不良的肿瘤患者常常表现高静息态能量消耗和高分解代谢。引发这些变化的因素主要包括两个方面:①肿瘤负荷;②由肿瘤引起的相关因素,包括炎症、神经内分泌紊乱、放化疗、肠道微生态紊乱以及食欲下降和摄入减少等。这些因素交杂在一起并通过尚未完全阐明的复杂机制而激活不同组织器官系统的能量消耗增加、分解代谢增强和合成代谢降低等,从而引起肿瘤患者的营养不良,甚至恶液质。由于引起肿瘤患者营养不良的因素和机制是复杂多样的,因此,肿瘤患者营养治疗也应该是针对性的综合治疗,即在营养不良综合评估和诊断基础上,进行有效综合营养干预和阻断高代谢的药物干预等。本文就肿瘤患者高能耗代谢及其相关机制进行综述。
许多肿瘤患者常见并发症是非自主性减重,最终导致恶液质。2020年《中国科学:生命科学》发表了常见恶性肿瘤营养状况与临床结局相关性研究(Investigation on Nutrition Status and Clinical Outcome of Common Cancers,INSCOC)团队的研究结果:中国肿瘤患者中、重度营养不良发生率为58%[1]。恶液质是近25%肿瘤患者死亡的直接原因。恶液质是由多因素引起并表现为骨骼肌持续减少、常规营养支持不能缓解和多器官功能受损的一种代谢紊乱综合征[2]。Ishida J[3]提出高代谢就是高静息态能量消耗(resting energy expenditure,REE),最常见于肿瘤恶液质患者。越来越多的研究表明,肿瘤代谢重编程和肿瘤相关的系统性炎症、内分泌紊乱、放化疗、心理应激等因素,通过复杂的机制引起肿瘤患者高耗能代谢状态。本文就肿瘤患者高能耗代谢及其相关机制进行综述。
1 肿瘤患者能量代谢变化
肿瘤恶液质患者体重下降实际上是能量摄取严重下降,能量消耗不断增加,或两者同时存在所导致的结果,也即能量代谢的严重失衡导致的一种临床恶性综合征。肿瘤患者能量代谢并不相同,约半数以上的肿瘤患者存在高代谢,研究显示肿瘤患者REE比健康对照平均高10%[4]。曹东兴等[5]研究发现70.6%肿瘤患者处于高代谢状态,23.7%处于正常代谢状态,而5.7%处于低代谢状态,经去脂体重(fat-free mass,FFM)及Harris-Benedict公式校正后恶性肿瘤患者总体处于高代谢状态。
REE增加程度取决于肿瘤类型、病理分期及进展程度等,见表1。比较不同类型肿瘤患者的能量消耗,发现食管癌、胃癌、胰腺癌、肺癌患者的REE/FFM显著高于结直肠癌患者和对照组。胃肠道或女性生殖系统来源的腹膜转移患者常伴有REE增加,肌肉质量下降和蛋白质分解代谢增加。Ⅳ期患者较早期肿瘤患者REE/FFM和测定REE(measured resting energy expenditure,mREE)/预测REE(predictedenergy expenditure,pREE)显著升高[6]。结肠癌患者REE水平受肿瘤分期及炎症程度影响较大[5]。Purcell SA等[7]对影响晚期结肠癌患者REE的决定因素进行了分析,发现只有肿瘤分期和全身炎症指标可预测REE的变化。

高代谢是早期肿瘤恶病质的决定因素。对390例成年肿瘤患者研究表明,49%患者出现高代谢并与负能量平衡,体重减轻、全身炎症和相关临床表现改变密切相关,多元线性回归分析显示高代谢、饮食和C反应蛋白(C-reaction protein,CRP)水平是体重丢失>5%(即恶液质状态)的独立危险因素[13]。同时也发现约52%患者体重丢失≤5%而出现高代谢状况[14]。肿瘤负荷对高代谢影响非常大,如膀胱癌患者进行根治性膀胱切除术后高代谢迅速恢复正常,但是体重下降的纠正较慢,在术后6个月时体重仍在下降[15]。这可能还与营养治疗等相关,及时提供足够的营养支持可以抵消高代谢、体重和肌肉损失[16]。因此,及时确定肿瘤患者高代谢,尤其是在体重丢失和出现相关症状之前,这对于临床治疗具有重要意义。
2 肿瘤患者高代谢机制
肿瘤患者高能耗代谢紊乱的因素是多样而复杂的,包括肿瘤恶性增殖和代谢重编程,这些因素交杂在一起通过复杂的机制激活不同器官系统(中枢神经系统、脂肪组织、胃肠系统、肝脏和肌肉),其能量代谢及机制见图1。肿瘤和宿主来源的细胞因子引起全身炎症,下丘脑炎症加剧了厌食症。此外,脂肪因子、肌肉细胞因子和脑源性厌食因子也影响骨骼肌和脂肪的消耗,并增加肿瘤微环境的炎症。组织器官间对话(cross talk)直接或间接影响组织代谢和恶液质的严重程度。炎症因子可增加白色脂肪组织的脂解作用,脂肪组织棕色化增加产热作用,促进肌肉消瘦。肌蛋白消耗会释放游离氨基酸,从而驱动肝脏的急性期反应(acute phase response,APR)。肿瘤相关的肠道微生物群异常会改变骨骼肌线粒体能量代谢,从而导致肿瘤患者的能量负平衡。在肿瘤骨转移情况下,破骨细胞活性增加导致骨基质转化生长因子-β(transforminggrowth factor-β,TGF-β)活化,从而促进肌肉消瘦并降低肌肉强度。胃肠道肿瘤或癌性肠梗阻的患者其能量物质的吸收常受到严重影响,高达83%的上消化道恶性肿瘤患者会发生体重减轻[17]。以下主要探讨引起肿瘤患者高代谢紊乱的内源性因素及机制。
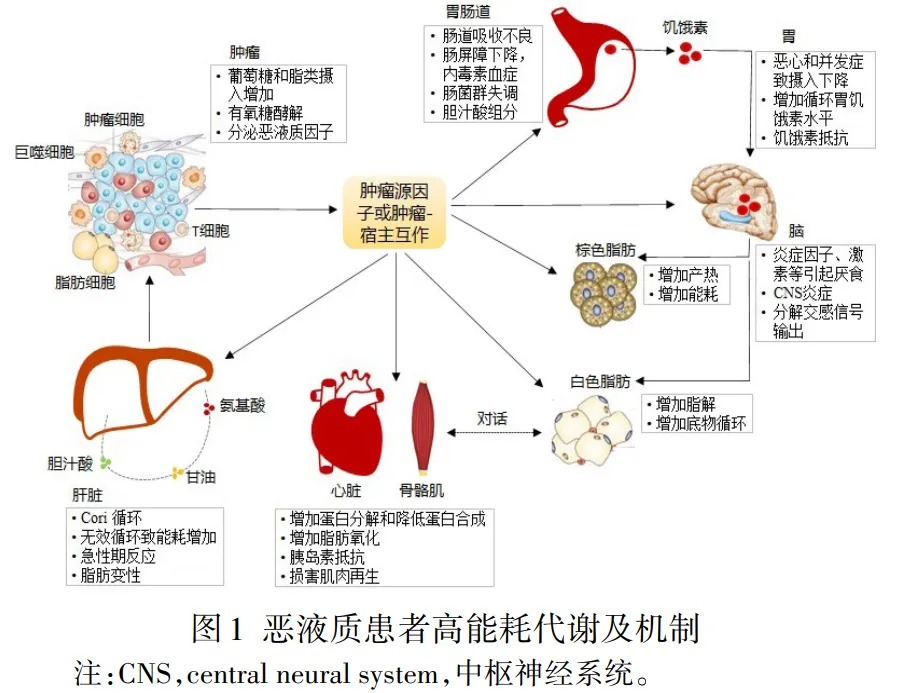
2.1 炎症 炎症因子通过多种机制在肿瘤患者尤其是恶液质阶段的REE中起着关键作用,见表2。高水平肿瘤坏死因子α(tumour necrosis factor-α,TNF-α)和白细胞介素-6(interleukin-6,IL-6)可引发高能量消耗和肌肉损失的主要炎症因子[18]。TNF-α可激活泛素E3连接酶-蛋白酶促进肌肉蛋白降解,是引起恶液质脂肪和骨骼肌消耗的主要细胞因子。一项回顾性队列研究显示,TNF基因的单核苷酸多态性(single nucleotide polymorphism,SNP)与恶液质患者体重减轻和骨骼肌指数降低有关[19]。动物模型显示表达TNF-α肿瘤细胞会导致恶液质,而不表达TNF-α的肿瘤细胞则不发生恶液质[18]。TNF-α还可抑制脂肪细胞和肌细胞分化,刺激脂肪分解,损害胰岛素信号,影响食物摄入,直接导致肌肉萎缩[20]。肿瘤患者骨骼肌中TNF-α部分通过激活相关蛋白激酶和IKKβ/NF-κB通路参与胰岛素抵抗,而NF-κB信号通路可刺激炎症因子和金属蛋白酶分泌增加等进一步促进肿瘤患者的肌肉减少和热量产生。TNF-α还可激活单核细胞趋化蛋白1(monocyte chemotactic protein 1,MCP-1)的表达,招募单核细胞进入脂肪组织而促发炎症。研究证明TNF-α还能激活细胞内果糖6-磷酸和果糖1,6-二磷酸之间的无效底物循环,形成能量消耗陷阱[21]。脑室内给药TNF-α可引起体重减轻和激活棕色脂肪组织,从而促进脂肪酸氧化分解并释放热能[22]。
其他细胞因子(IL-1β、IL-6、IL-10和IFN-γ)基因SNP也与胰腺癌和胃癌恶液质相关[23],循环IL-6水平与体重减轻和生存相关[24],IL-6可与神经元细胞膜表面gp130受体结合,参与患者厌食症的发生,还可通过与肝细胞膜表面gp130受体结合而激活下游JAK/STAT通路,进而诱导急性期蛋白的转录和合成[25,26]。IFN-γ升高与肿瘤发展过程中的厌食、胰岛素抵抗、脂肪降解和体重减轻密切相关。总之,慢性炎症是影响大多数组织器官能量消耗进而导致恶液质发生和发展的主要因素之一。
2.2 特异性代谢因子 肿瘤来源特异性代谢因子,包括脂肪动员因子(lipid-mobilizing factor,LMF)和蛋白水解诱导因子(proteolysis-inducing factor,PIF)也是引发肿瘤患者高能耗代谢紊乱的重要因子,见表2。

2.2.1脂肪动员因子 LMF是一种锌-α2-糖蛋白除了肿瘤细胞外,肺、心、棕色脂肪(brownadipose tissue,BAT)和白色脂肪组织(white adipose tissue,WAT)也表达LMF/ZAG,脂肪细胞分泌的LMF/ZAG可能通过自分泌或旁分泌方式作用于局部脂肪组织分解脂肪。在脂肪组织下降60%的荷瘤鼠模型上,WAT的ZAGmRNA表达水平增加10倍,BAT增加3倍,而同时发现WAT的瘦素下降了33倍。当体重下降达24%时,WAT和BAT中ZAG蛋白质水平分别增加了10倍和20倍。这表明ZAG是一个重要脂肪细胞因子,并在脂肪代谢和恶液质发生过程中发挥着重要作用。在LMF/ZAG主要通过经典依赖GTP的腺苷酸环化酶-cAMP通路激活激素敏感脂肪酶(hormone-sensitivelipase,HSL)分解脂肪。LMF/ZAG促进脂肪动员同时还加强脂类的氧化分解,其机制可能通过激活肾上腺素能受体β3-腺苷酸环化酶-cAMP通路而促进解偶联蛋白(uncoupingprotein,UCP)1和UCP2表达有关;LMF/ZAG还以剂量依赖方式并通过丝裂原激活的蛋白激酶(mitogen-activated protein kinase,MAPK)通路促进UCP3表达。小鼠模型研究发现LMF/ZAG还增加脑、心、BAT和骨骼肌利用葡萄糖,降低血糖,同时以依赖GTP方式激活腺苷酸环化酶而消耗肝糖原。多种机制可以调节ZAG的表达:关于人脂肪细胞的研究发现过氧化物酶体增殖物激活受体g(peroxisome proliferator activated receptor g,PPARg)激动剂罗格列酮可上调ZAG表达3倍,TNF-α可上调ZAG 4倍,同时ZAG还受到肾上腺素能受体β3激动剂BRL37344和糖皮质激素调节。糖皮质激素拮抗剂RU38486可以明显减轻恶液质患者的体重下降和WAT的ZAG水平;恶液质鼠血浆皮质醇水平与体重丢失成正比,营养不良的恶液质患者尿液中皮质醇明显增加,还有儿茶酚胺类增加也
恶液质患者 腹腔广泛肿瘤种植
恶液质患者 病人非常消瘦
与恶液质ZAG水平有关。细胞实验发现ω-3脂肪酸二十碳五烯酸(eicosapentaenoicacid,EPA)可以减轻由地塞米松诱导人脂肪细胞的脂肪降解和ZAG 表达,这可能与EPA治疗恶液质有关。
2.2.2蛋白水解诱导因子 PIF是一种分子量为24 kDa的硫酸化糖蛋白,通过作用于肌肉和肝脏细胞膜上的PIF受体,激活NF-κB通路而抑制蛋白质合成和增加蛋白质降解[30,31]。从MAC16肿瘤中分离的PIF或胰腺癌恶液(zinc-a2-glycoprotein,ZAG)可增强脂肪细胞对脂解刺激的敏感性,促进脂肪分解和增加产热,研究证实将LMF/ZAG注入小鼠体内后引发脂肪动员、厌食和恶液质[27,28]。质患者尿液中分离的PIF都可迅速降低小鼠体重,24 h内体重下降达10%,并且专一性消耗瘦肉组织,小鼠腓肠肌下降64%,比目鱼肌下降17%,而心和肾没有影响,肝脏重量增加。PIF特异性增加腓肠肌泛素、E214k和C9蛋白酶体亚基表达,即通过增加泛素-蛋白酶体通路分子表达促进骨骼肌蛋白降解的。研究揭示PIF作用需要通过激活转录因子NF-κB信号通路。IKKβ/NF-κB对于诱导泛素-蛋白酶体通路激活发挥重要作用,通过小鼠肌肉专一性表达活性IKKβ激活NF-κB可引起类似临床恶液质的肌肉消耗,包括E3连接酶MURF1表达升高3.3倍,蛋白酶体C2和C9亚基表达升高2.4~2.8倍;激活NF-κB还可抑制肌源性转录因子MyoD表达,导致肌球蛋白合成下降。白藜芦醇通过抑制NF-κB可明显减轻荷瘤小鼠恶液质状况,包括能明显缓解体重下降和肌肉蛋白降解等。这进一步验证了NF-κB激活对于恶液质肌肉萎缩中的重要意义。
2.3 肠道微生态 肠道菌群是机体的一个新的动态功能器官,与机体消化吸收、免疫、神经内分泌,以及机体代谢稳态等密切相关。肿瘤患者肠道菌群常常发生紊乱,通过影响肠屏障和肠漏引起机体内毒素血症和炎症,从而影响宿主营养素消化和吸收、代谢效率和能量消耗。结肠黏膜细胞主要依赖肠道菌群产生的短链脂肪酸作为能源,每日提供10%能量需求。肠道菌群紊乱导致从短链脂肪酸氧化代谢转变为糖酵解,导致宿主细胞ATP不足[32]。膳食补充丁酸盐可增加小鼠的能量消耗和线粒体功能[33]。研究表明菌群-肠道-肌肉轴与肌肉消耗有关,选择性调节乳杆菌属可影响小鼠白血病恶液质模型肌肉萎缩和炎症标志物水平,而当恢复乳酸菌水平后可减轻恶液质小鼠肌肉萎缩程度[34]。通过合生元制剂恢复肠道微生物特征后成功治疗恶液质的肌肉萎缩和体重减轻。因此,合生元或菌群移植可能是未来恶液质治疗的潜在方法之一[35]。
2.4 内分泌紊乱 肿瘤患者常常存在明显的内分泌紊乱,主要表现为合成激素减少如胰岛素分泌减少和抵抗;同时分解激素如糖皮质激素、儿茶酚胺类和血管紧张素等分泌增加。
糖皮质激素由于其有效缓解恶液质症状(改善食欲、增加食物摄入和减轻不适感觉等)成为了恶液质的辅助用药,因为糖皮质激素可通过上调泛素-蛋白酶体通路引起肌肉萎缩[36]。所以只能限制在肿瘤终末期的短期应用。血管紧张素Ⅱ(angiotensin Ⅱ,ANG Ⅱ)也是一种可以促进骨骼肌蛋白分解的重要分子,临床研究显示用血管紧张素转化酶(angiotensin converting enzyme,ACE)治疗充血性心力衰竭时可以增加恶液质患者皮下脂肪和肌肉,而注入ANG Ⅱ可引起小鼠体重明显下降,并以瘦肉组织丢失为主。ANG Ⅱ与PIF类似机制通过蛋白激酶R(protein kinase R,PKR)激活泛素-蛋白酶体通路抑制蛋白质合成和促进蛋白质降解,以及通过蛋白激酶C(protein kinase C,PKC)➝NADPH氧化酶➝活性氧(reactiveoxygen species,ROS)➝NF-κB。而胰岛素样生长因子-1(insulin-likegrowth factor-1,IGF-1)可对抗ANG Ⅱ作用,通过诱导蛋白磷酸酶1(protein phosphatase 1,PP1)表达,使PKR脱磷酸而抑制NF-κB和泛素-蛋白酶体通路激活,同时通过降低真核生物起始因子(eukaryotic initiation factor 2,eIF2)的α亚基磷酸化来减轻蛋白质合成的抑制作用[36,37]。
神经内分泌肽和神经递质通过自主神经系统和内分泌系统作用于靶器官,参与食欲、肠蠕动和胃酸分泌以及脂肪和葡萄糖代谢,从而达到对能量的动态调节。瘦素通过下丘脑神经肽降低食欲并增加能量消耗。饥饿状态或体内脂肪减少时会导致瘦素减少,使体内能量处于正平衡状态。胃饥饿素、神经肽Y和其他食欲刺激性神经肽的增加,促肾上腺皮质激素释放因子和黑皮质素等激素的活性降低介导了这种代偿反应。而肿瘤会产生诱导或模仿瘦素过多的负反馈信号,导致补偿反应无法进行,引起持续厌食(食欲不振)和恶液质(肌肉消瘦和体重失控)[2]。另外,在肿瘤患者血浆和脑中发现色氨酸和5-羟色胺(5-hydroxytryptamine,5-HT)水平显著升高。5-HT可能与饱腹感相关,是导致厌食症及恶液质的重要效应因子之一。在恶液质小鼠中儿茶酚胺信号传导增强,普萘洛尔阻断β-肾上腺素可以阻止产热增加[38]。近50%转移性肿瘤的男性患者的睾丸雄激素降低,肌肉量和性功能降低。睾丸雄激素降低和促分解激素增加是导致恶液质相关骨骼肌分解的重要因素[39]。
最近的一项研究表明甲状旁腺激素及其受体在晚期慢性肾脏疾病的肌肉萎缩和恶液质发生中发挥新的作用[40]。而最新动物研究发现路易斯肺癌通过细胞外囊泡释放甲状旁腺激素相关蛋白(parathyroid hormone related protein,PTHrP)诱导肿瘤恶液质患者的脂肪分解和脂肪组织棕色化,同时还可引起肌肉分解萎缩[41]。
2.5 底物无效循环 慢性消耗性疾病,如肿瘤引起的恶病质、败血症和烧伤等具有许多相似的代谢表型,表明不同的病因可能触发身体消耗的共同下游事件,即生物化学上能量消耗的无效循环反应,无效循环是指代谢通路如糖酵解和糖异生代谢途径大多是可逆反应,但有3个不可逆反应并有不同酶催化其单向反应,当2种酶活性相等时,则不能将代谢向前推进,结果仅是ATP不断分解消耗,因而称之为无效循环(futile cycle),包括葡萄糖和6-磷酸葡萄糖、6-磷酸果糖和1,6-二磷酸果糖、乳酸和葡萄糖之间的Cori循环、脂肪分解和再酯化,以及肌酸激酶依赖的肌酸和磷酸肌酸等无效循环[38,42],不受控制的无效循环反应会大量消耗能量,见图2。
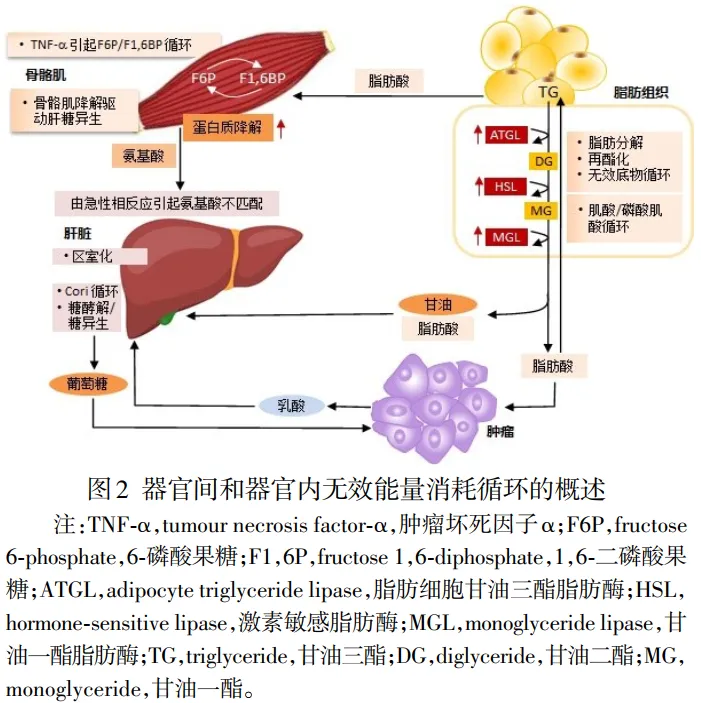
2.5.1肿瘤与肝脏之间类Cori循环 研究发现肿瘤细胞糖酵解能力是正常细胞的20~30倍,最高可达到正常的200倍。因此,肿瘤组织不断摄取葡萄糖而大量排出乳酸,进入肝脏糖异生,肝脏产生的葡萄糖又大量被肿瘤细胞摄取。这样在肿瘤与患者肝脏之间形成类似Cori循环,即葡萄糖-乳酸循环。从糖酵解能量产生和肝脏糖异生消耗能量来看是得不偿失的:肝脏2 mol乳酸合成1 mol葡萄糖要消耗6 mol ATP,这样就增加了葡萄糖和ATP的无效消耗,葡萄糖利用效率明显下降。研究证实转移性结直肠癌患者比对照组具有较高效率的葡萄糖-乳酸循环,这样通过Cori循环消耗能量达到300kcal/d[43]。另外,肿瘤乳酸水平可能与肿瘤转移和复发呈正相关,而与患者生成呈负相关。
2.5.2肌肉与肝脏之间丙氨酸-葡萄糖循环 由于肿瘤患者存在胰岛素分泌不足或胰岛素抵抗等导致肝脏糖异生通路异常活跃,同时大量糖异生原料(甘油和生糖氨基酸等)进入肝脏进行糖异生而消耗大量能量,如肿瘤患者,尤其是恶液质状态下,脂肪组织和骨骼肌分解加强,产生大量的甘油和氨基酸进入肝脏糖异生。其中肝脏与肌肉之间的丙氨酸-葡萄糖循环也是一条重要的循环耗能通路,2 mol丙氨酸异生1 mol葡萄糖时消耗6 mol ATP,同时丙氨酸脱氨产生2 mol NH3将会进入鸟氨酸循环通路合成1 mol尿素消耗4 mol ATP,这表明2 mol丙氨酸通过丙氨酸-葡萄糖循环和鸟氨酸循环共消耗10 mol ATP。因此,肿瘤患者出现骨骼肌分解萎缩时将导致丙氨酸-葡萄糖循环增强进而增加耗能[17]。
2.5.3其他无效循环 由于肿瘤患者早期就存在脂肪分解,释放大量甘油和游离脂肪酸(free fatty acid,FFA),FFA除了氧化分解外还可以再酯化生成三脂酰甘油(triacylglycerol,TAG),这也就是TAG-FFA底物循环。在荷瘤小鼠模型中发现TAG-FFA循环率显著高于非荷瘤小鼠组。另一个无效循环是线粒体的质子循环,由于肿瘤患者炎症等因素导致UCP表达升高,导致线粒体解偶联的无效质子循环增强进而增加能量消耗[17]。
2.6 白色脂肪棕色化 某些恶液质患者高REE可能与BAT、白色脂肪棕色化以及肌肉组织产热作用增强有关,并且发现肿瘤恶液质早期就发生白色脂肪棕色化[44]。一般来说成人只有很少的BAT,但是尸检结果显示80%恶液质患者肾上腺周围存在大量BAT[45]。BAT和肌肉组织产热源于其表达大量的UCP,UCP可使线粒体内氧化磷酸化产生的质子梯度转化为热能释放,与能量消耗相关UCPs主要包括UCP-1、UCP-2和UCP-3。研究发现,恶液质患者和动物模型脂肪组织都高表达UCP-1。骨骼肌过表达UCP‑3转基因小鼠食欲旺盛而体重低于对照动物,并且脂肪组织明显减少。当荷瘤小鼠(MAC16结肠腺癌)体重丢失达24%时,棕色脂肪组织UCP-1 mRNA水平明显升高,而骨骼肌UCP-2和UCP-3 mRNA水平也明显升高;而在易产生恶液质的荷瘤小鼠(吉田腹水肝细胞癌)也出现骨骼肌的UCP-2和-3mRNA水平明显升高,且骨骼肌UCP-3 mRNA水平直接与血清FFA水平相关,在大鼠恶液质模型中,UCP增加同时伴有2倍以上的血液FFA升高。一种噻唑烷二酮类药物类降糖药曲格列酮可以选择性激活PPARg,而显著降低小鼠骨骼肌UCP-2和UCP-3 mRNA水平,这提示PPARg配体可降低恶液质能量消耗。因此,曲格列酮类药可能对于肿瘤患者有一定应用价值。
有证据表明某些细胞因子和LMF可以增加BAT和骨骼肌中UCP表达。慢性炎症和IL-6可促进WAT中UCP1的表达,而抑制炎症或β-肾上腺素能阻滞剂可降低WAT褐变和改善恶液质的严重程度。另外Kir S等[45]在研究lewis肺癌恶液质小鼠模型时发现肿瘤来源PTHrP可激活脂肪组织UCP1表达,促进脂肪棕色化和产热,而中和PTHrP可以阻止脂肪组织棕色化,还可抑制肌肉质量和力量的损失,以及恶液质症状得到控制或改善。这表明PTHrP在恶病质发生和发展中发挥多方面作用。因此,抑制白色脂肪棕色化可能是改善肿瘤恶液质的一种潜在治疗策略。
3 小结
目前尚没有完全清楚高代谢是否可能是晚期肿瘤患者的治疗靶点。然而,一些研究,如Ma YJ等[46]研究给予ω-3多不饱和脂肪酸可显著降低REE,显著提高胰腺癌患者的总生存率。一些药物如饥饿素激动剂、选择性雄激素受体分子、醋酸甲羟孕酮、激活素受体拮抗剂、肌钙蛋白抑制剂[47],以及体育锻炼都有治疗或预防肌肉消耗的作用。同时一些针对高代谢的药物干预,如抗环氧化酶-2或β-肾上腺素能受体阻滞剂尚未在大型肿瘤患者试验中进行评估。一项对烧伤后高代谢期研究的meta分析显示,普萘洛尔阻断肾上腺素受体能著降低REE,改善外周瘦体组织量[48]。还有目前尚没有能有效阻断器官间和器官内高耗能的无效底物循环阻滞剂的相关基础研究和临床应用研究。这些可能都是潜在治疗肿瘤恶液质的候选药物和方法。同时,亟需进一步阐明肿瘤患者高代谢的病理生理学及生化代谢机制,同时需要开展针对不同肿瘤类型和不同病理阶段的更大样本量肿瘤患者的临床验证研究。总之,导致肿瘤患者营养不良和恶液质的因素和机制是复杂多样的,肿瘤恶液质治疗也应该是个体化的综合治疗,在营养不良综合评估和诊断基础上,进行有效营养干预和靶向高代谢药物干预等。
本文为转载文章,如有侵权请联系作者删除。本文仅供健康科普使用,不能做为诊断、治疗的依据,请谨慎参阅
评论