


 三甲
三甲
医学科普——与鳃器异常发育相关的颅面综合征
医学科普——与鳃器异常发育相关的颅面综合征
广东省人民医院&广东省医学科学院
耳鼻咽喉头颈外科
陈良嗣头颈腺体、先天疾病及儿童头颈外科工作室
鳃器包括鳃弓、鳃裂、鳃膜和咽囊,胚胎早期,它们共同参与颜面颈部及相关组织器官的发生发展。
鳃裂、鳃膜和咽囊的单纯性闭合异常或退化不全,可以导致一系列鳃源性畸形。鳃器的异常衍变或发育障碍还可以导致或伴随某些严重的颅面相关综合征。本篇尝试简要阐述。
【Treacher-Collins Syndrome,TCS(特雷彻·柯林斯综合征)】
又称Franceschetti综合征、鸟面综合征、鱼面综合征、下颌面骨发育不良综合征,是胚胎早期第一、第二鳃弓异常发育所致的先天颅面复合畸形,1900年,英国外科及眼科医生Edward Treacher Collins首次描述了TCS,主要累及中、下面部,包括骨组织和软组织畸形。
发病率:极为罕见,新生儿发病率为万分之0.2-0.4,
遗传:40%患者有家族史,呈常染色体显性遗传;60%患者表现为新生突变,涉及的基因包括TCOF1、POLR1C或POLR1D。
临床表现(附图1):主要特征为颅面骨发育不全(特别是颧骨、下颌骨),双眼外眦下移、巨口、面部瘘管、外耳畸形等。

- 眼:下眼睑缺损,内侧睫毛发育不全,外眦下移,睫毛缺失。视力损失,伴有斜视,屈光不正。
- 耳:耳廓畸形伴有外耳道闭锁,一般是对称性,多数表现为耳廓和中耳腔畸形,偶伴有内耳畸形。存在中重度的传导性听力下降。
- 颌面:颧骨和下颌骨发育不全,伴牙齿咬合不良,下巴短小。
- 腭裂:腭裂会引起通气困难,造成呼吸困难。
- 口腔畸形:60%患者会出现牙齿畸形,其中包括,牙齿发育不全、牙釉质发育畸形、第一磨牙异位。咬合不良,出现摄食困难和难以闭口。
- 其他表现:鼻畸形,弓状腭,上眼睑缺损,眼距过宽,鼻后孔闭锁,巨口,耳部头发移位。
- 智力:多正常。
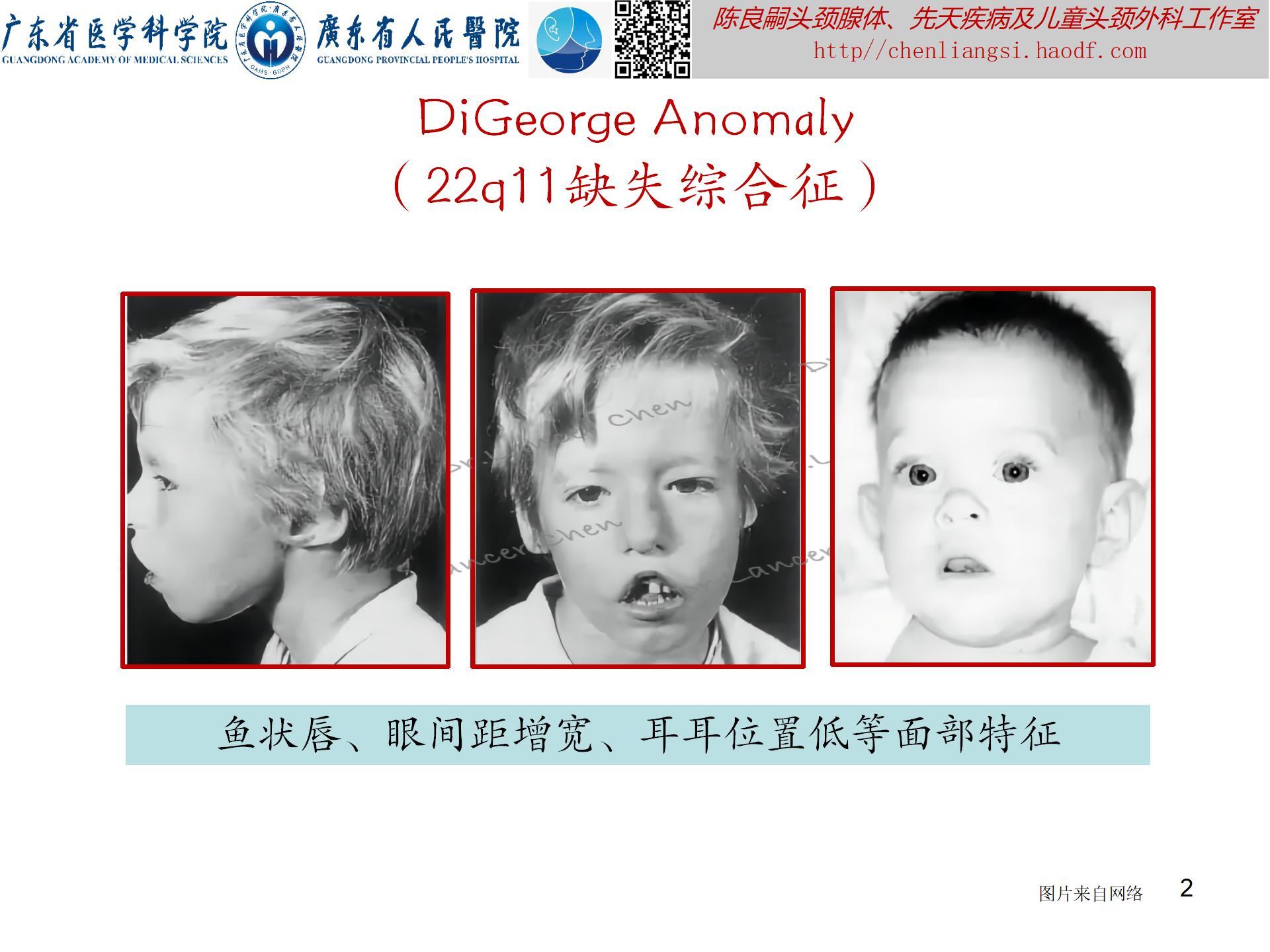
【DiGeorge Anomaly 】
DiGeorge异常(DiGeorge anomaly)即胸腺缺陷(thymic defects),曾称为DiGeorge综合征(DiGeorge syndrome, DGS)或先天性胸腺发育不良,是一种第三、四对咽囊发育障碍导致的先天性异常伴免疫缺陷性疾病,又称“第三、第四咽囊综合征”。1965年,美国医生Angelo DiGeorge首先描述并报道命名。临床包括胸腺发育不良、低钙血症、先天性心脏病(心脏圆锥动脉干畸形)和面部畸形等一组疾病,根据免疫损伤程度,分完全型(胸腺缺陷)和部分型(胸腺部分损害)。
发病率:约为1∶2000~1∶4000。
遗传:7%的病例有遗传背景,呈常染色体显性遗传;约85%的病例为散发,与染色体22q11.2杂合性缺失有关,也称“22q11缺失综合征”。
临床表现(附图2):多样,所有症状的首字母缩略词为CATCH-22(cardiac,abnormal facies,thymic hypoplasia,cleft palate,and hypocalcemia resulting from 22q11 deletion,22q11缺失导致的心脏畸形、面容异常、胸腺发育不良、腭裂以及低钙血症)。
- 心血管系统:约85%患者有心脏缺陷。最常见畸形有:法洛四联症(TOF)(25%);动脉单干;主动脉弓离断(15%);室间隔缺损(VSD),通常是膜周部(15%);动脉导管未闭(9%);及单纯的主动脉弓畸形如右锁骨下动脉迷走(5%)。较少见的包括肺动脉狭窄(PS)、房间隔缺损(ASD)、房室共道及完全性大动脉转位。
- 特殊面容及体征:眼距过宽、眼裂上斜、双眼皮、鼻尖圆钝、小下颌畸形、像鱼嘴样外形的短人中、高腭弓、低耳位(常伴耳廓畸形)、锥形手指。
- 免疫:胸腺发育不良或胸腺缺如造成轻度到中度T细胞数量降低;偶尔可见体液免疫缺陷,包括IgA缺乏(10%左右)。根据胸腺受累程度不同分为完全性和部分性DiGeorge综合征。
- 腭:腭部畸形常见(70%~80%),伴有说话困难和喂养障碍。偶可存在双侧唇腭裂。腭咽闭合功能不全可造成说话延迟和鼻音过重。
- 代谢:甲状旁腺功能减退引起低钙血症(60%),可表现为抽搐。
- 反复感染:常见,主要为细胞免疫缺陷所致,且为晚期死亡的重要原因。
- 全身:婴儿期身材短小、精神发育迟缓、肌张力降低是常见的表现。偶见精神疾病(如精神分裂症或双向性精神障碍)。70%~90%的DiGeorge综合征患儿可出现发育落后和学习困难。
【Pierre Robin Sequence,PRS(皮尔罗宾氏征)】
皮尔罗宾氏征(Pierre Robin Syndrome,PRS)是由法国牙医Pierre Robin于1933年命名。又称Rabin综合征、腭裂-小颌畸形-舌下垂综合征、小颌-舌根下沉-吸气性气道阻塞综合征等。该畸形可单独存在,也可以是某些多发、复杂先天性畸形的一部分,如Stickler 综合征、velocardiofacial综合征及Treacher-Collins综合征。当Pierre Robin序列征与其他综合征无关的时候,其称为非综合征性Pierre Robin序列征。
发病率:在1/20000-1/8500之间。
遗传:可能为常染色体隐性遗传(AR)或X染色体隐性遗传(XR)。
临床表现(附图3):PRS是一组以小颌畸形、舌后坠、腭裂及上呼吸道梗阻为特征的先天性畸形。
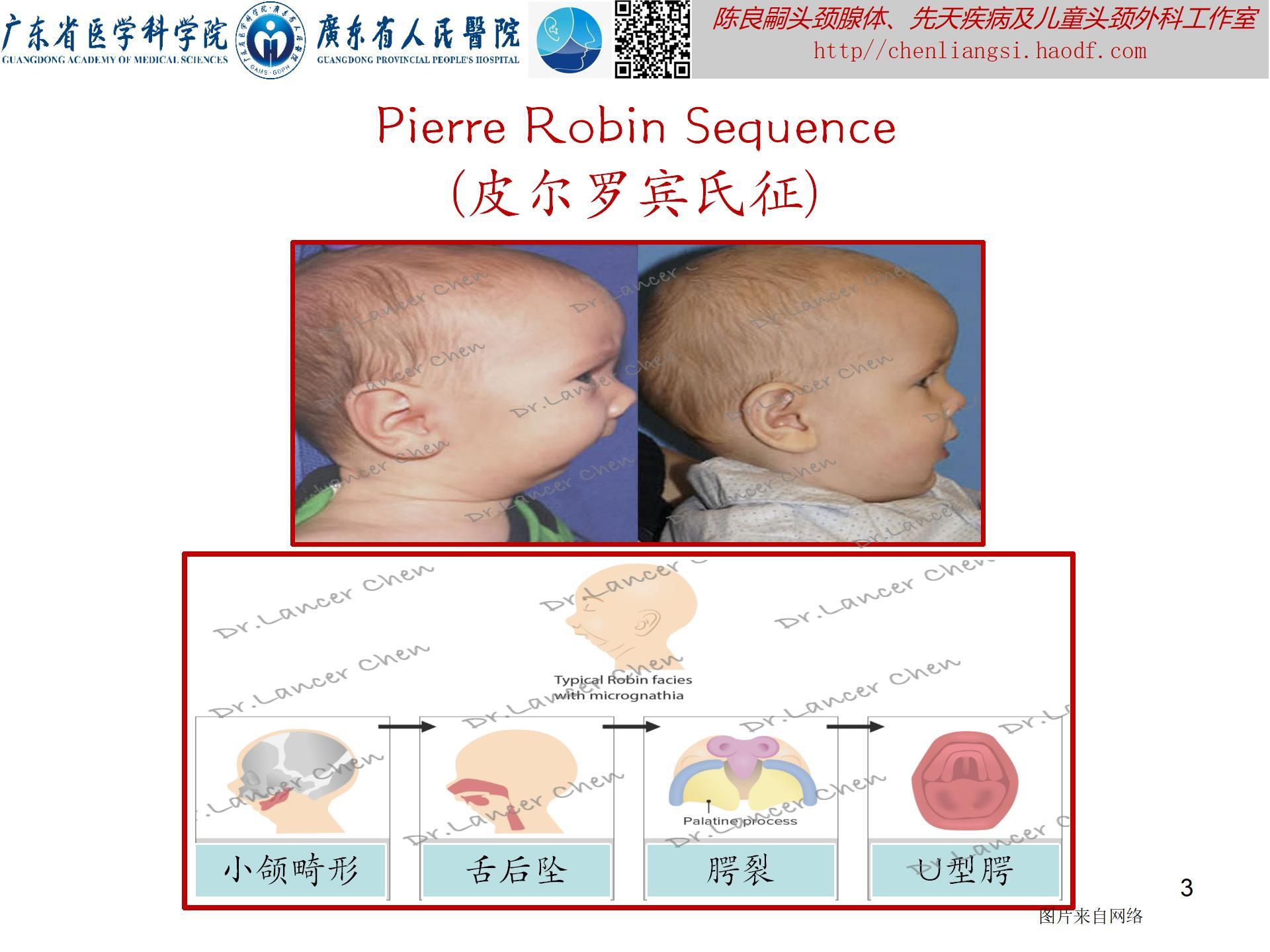
- 由下颌骨发育不良或异常后缩引发的一系列后续的结构变异,其中下颌骨发育不良以颏部最为明显,呈现特殊的面容。
- 腭裂的发生率约为90%,多为不完全性腭裂。
- 多数PRS患儿都伴有不同程度的喂养困难、营养不良、呼吸困难甚至睡眠窒息。
- 部分患儿可伴有其他系统畸形如心血管畸形、多指畸形、小脑发育不全、耳部畸形等。
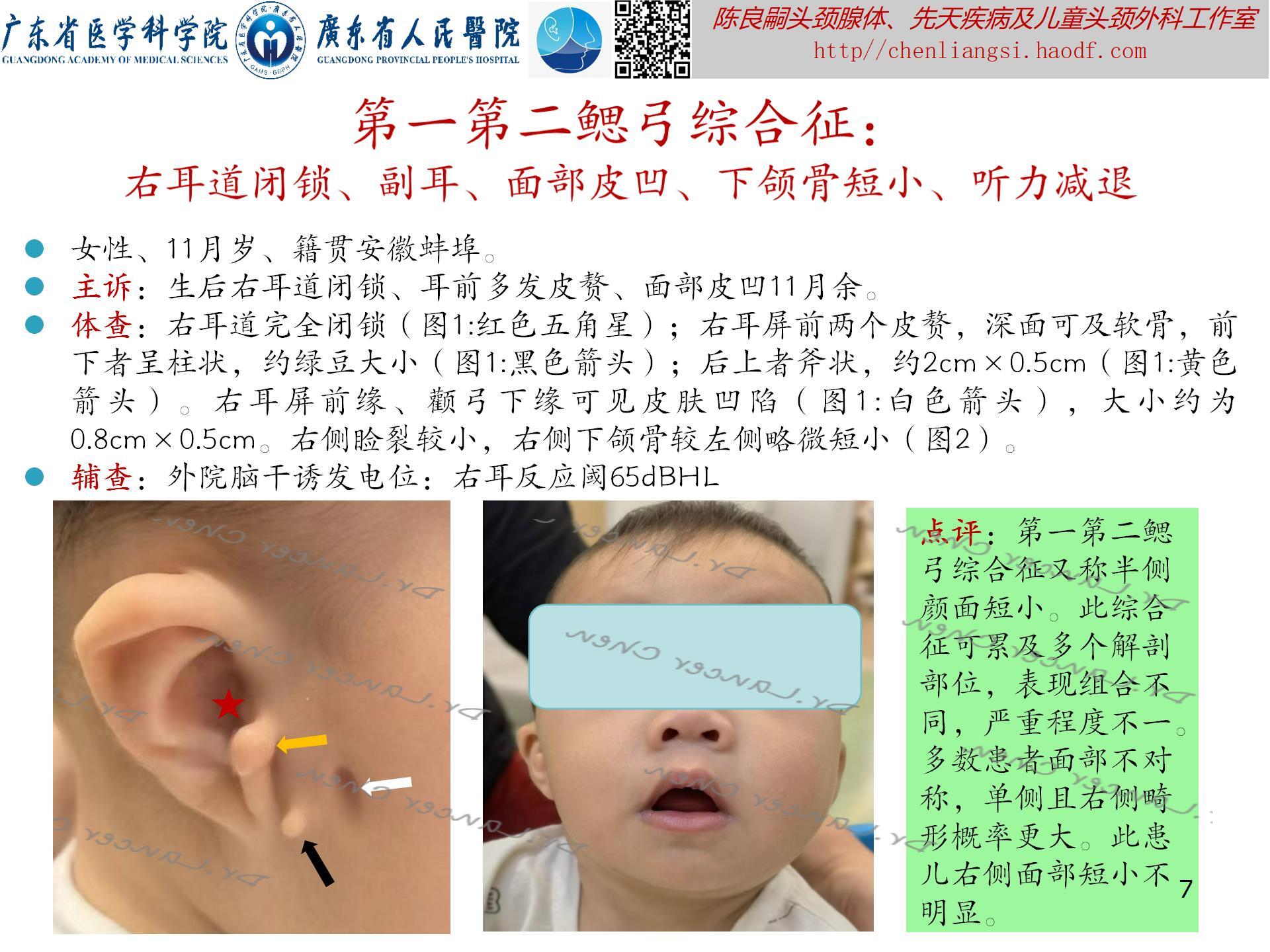
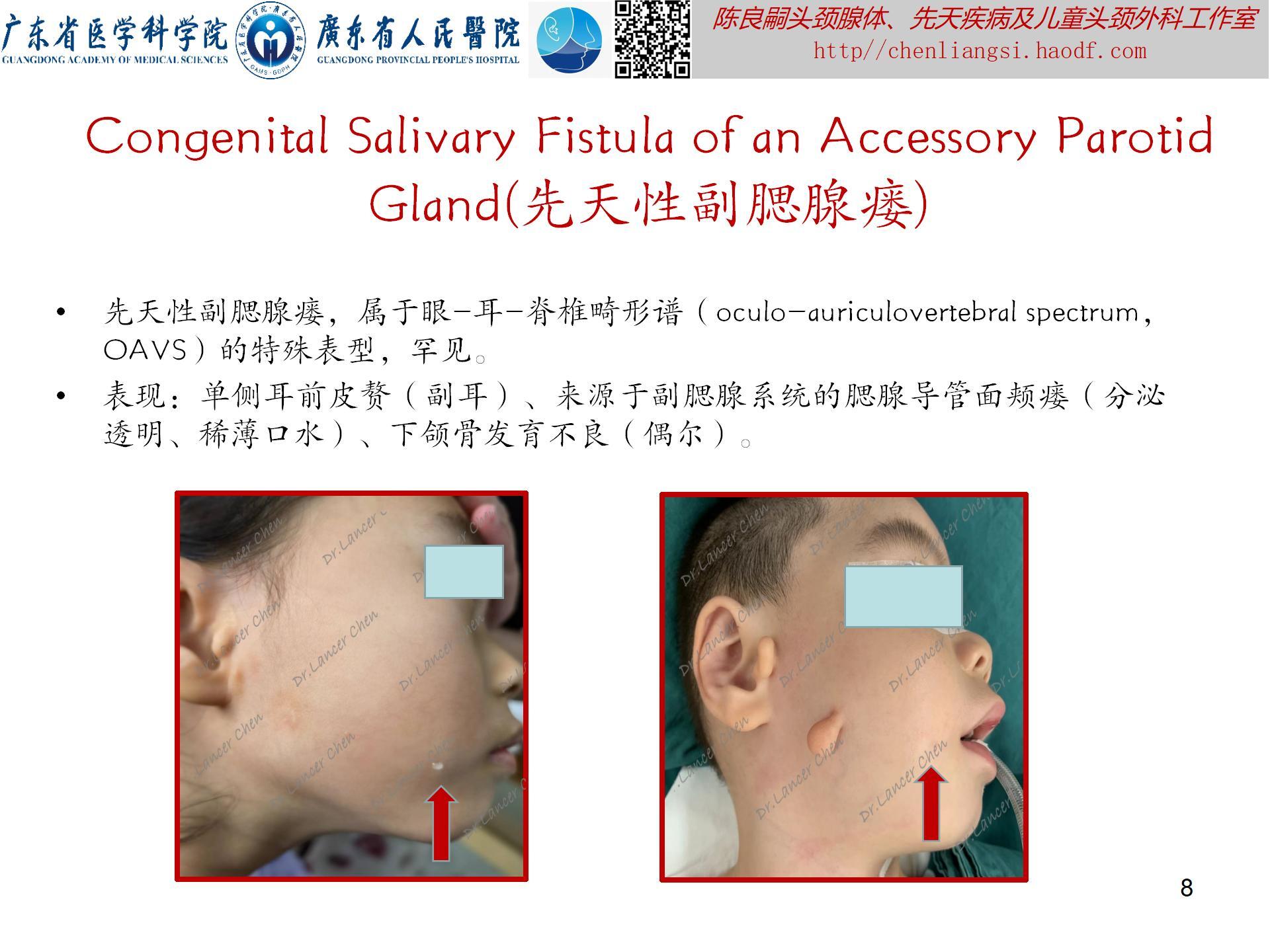
【oculoauriculovertebral spectrum,OAVS(眼-耳-脊椎畸形谱)】
是一种以眼、耳及颜面和脊柱畸形为主要临床症状的先天性症候群。原称为“眼-耳-脊椎畸形发育不良”(oculoauriculovertebral dysplasia),因表型广泛,故有许多术语定义此类畸形,如:半侧颜面短小(Hemifacial Microsomia),单侧颅面发育短小(unilateral craniofacial microsomia,UCM)、第一第二鳃弓综合征,面-耳-脊椎综合征、Goldenhar综合征等。因上述每种畸形综合征都有表型重合,故1989年Cohen等人重新定义其为“眼-耳-脊椎畸形谱”。可能与胚胎时期第一、第二鳃弓以及眼和脊柱的血管异常引起的发育畸形。
发生率:出生活婴的1/3500-1/26550,是仅次于唇腭裂的、最常见的先天性颅面畸形。
遗传:OAVS属于单基因疾病。已知的致病基因有MYT1、ZYG11B、EYA3、 SF3B2。约12%OAVS患者有家族遗传史,遗传方式:常染色体显性遗传、常染色体隐性遗传。 大多OAVS患者为散发病例。
临床表现(附图4-8):此综合征可累及多个解剖部位,表现组合不同,严重程度不一。多数患者面部不对称,单侧且右侧畸形概率更大。
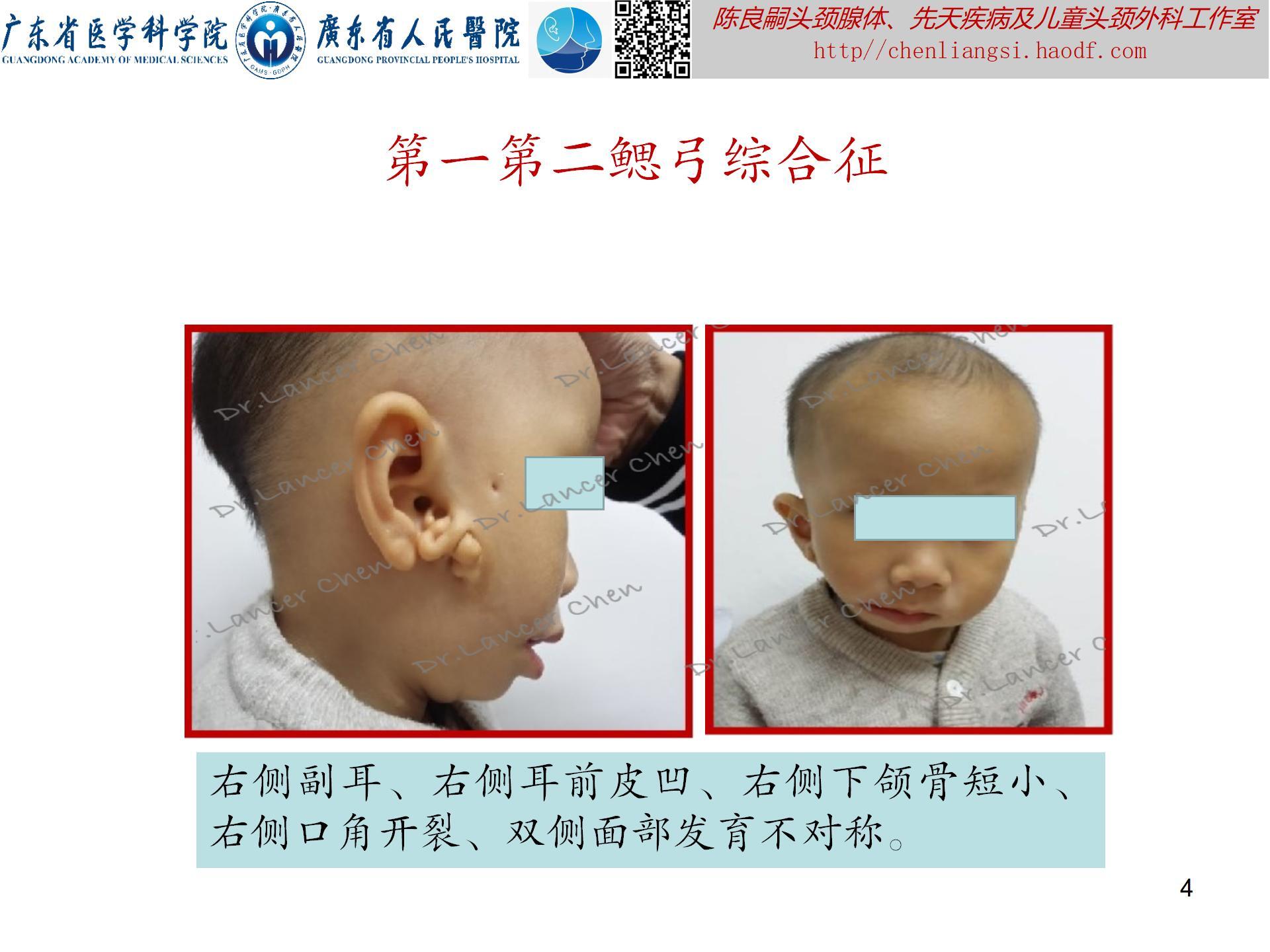
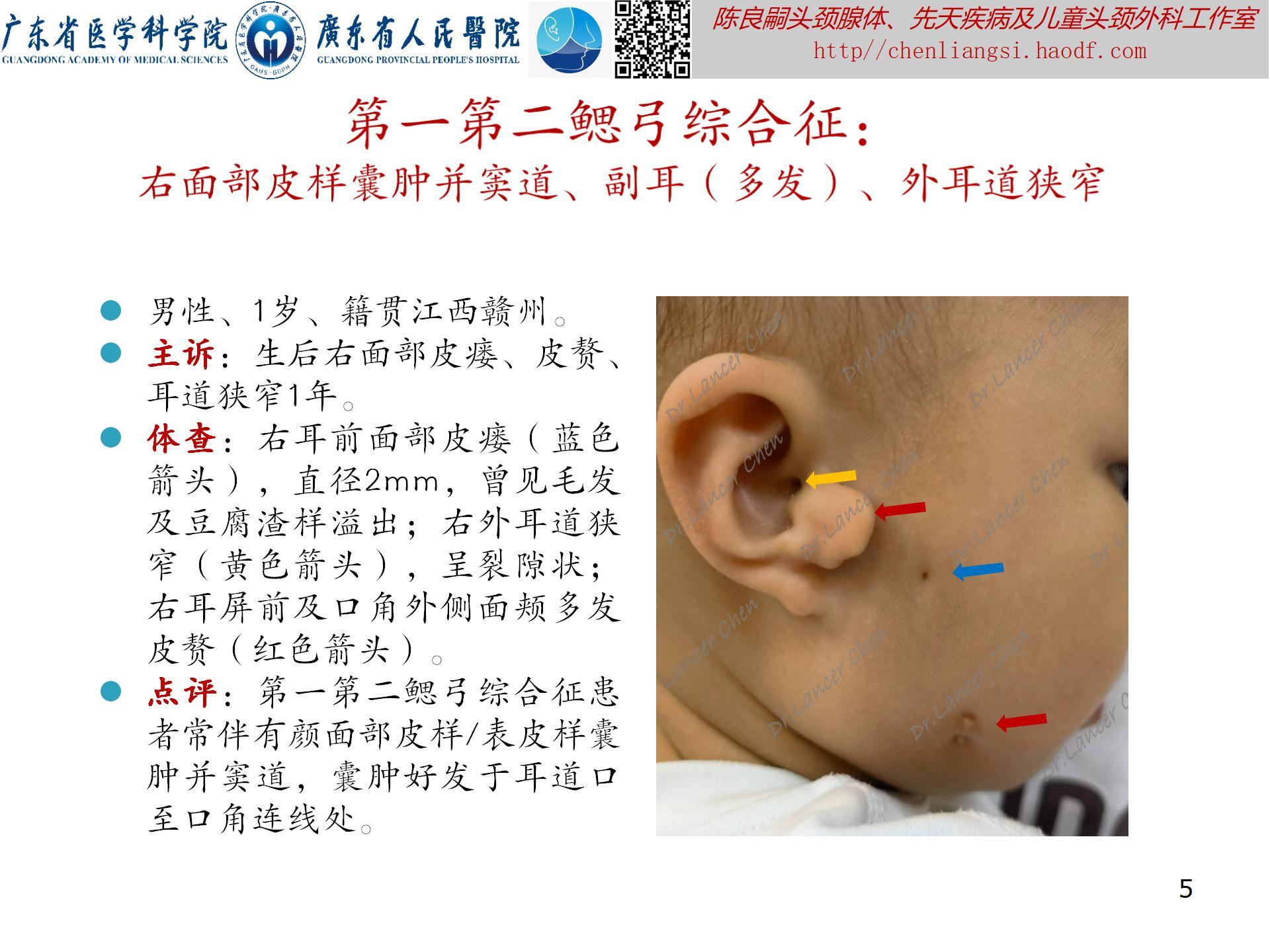
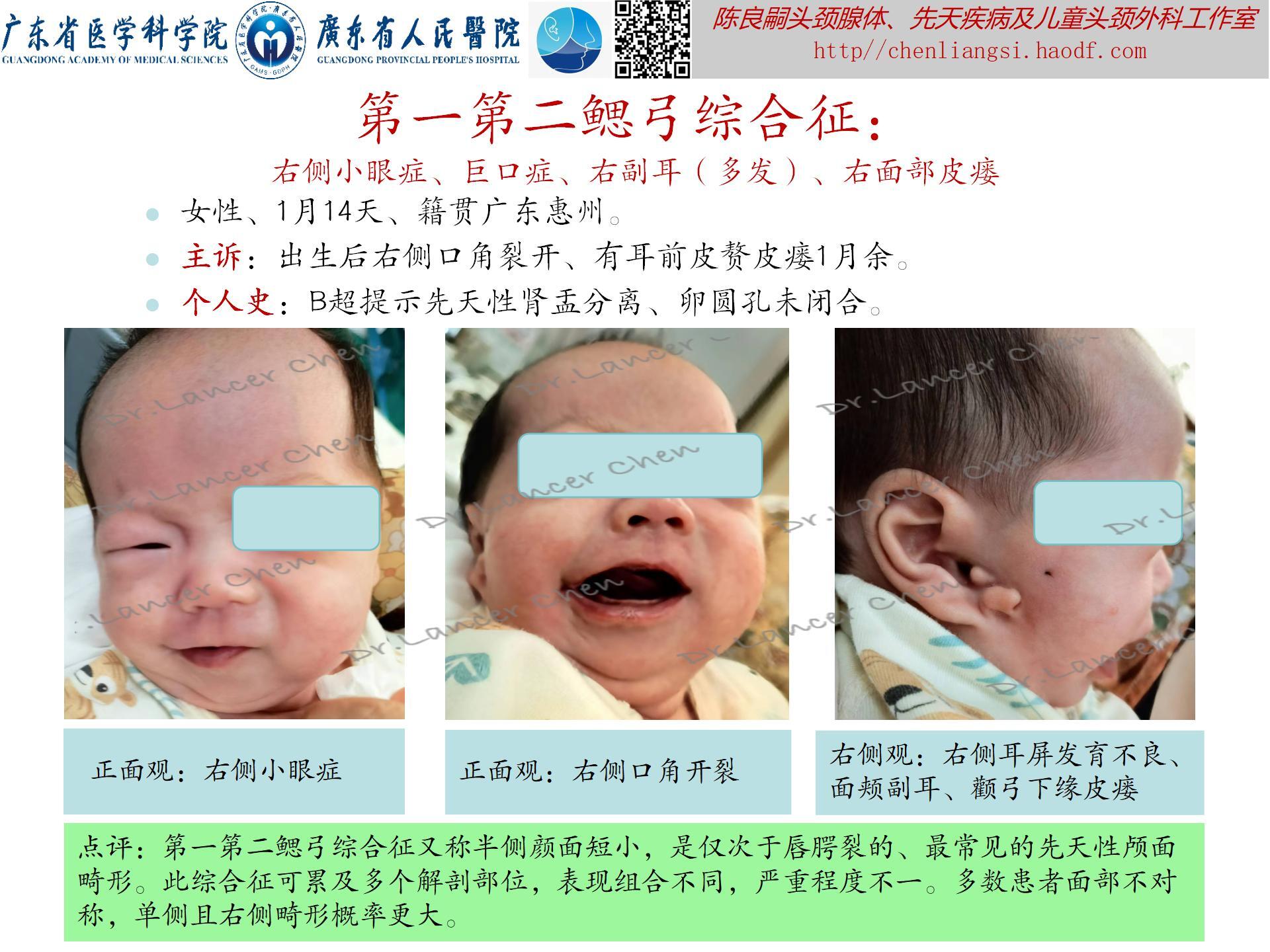
- 眼部:眼球表皮样囊肿/脂肪样囊肿;上眼睑缺损;斜视;小眼症;泪管闭锁/狭窄。
- 耳部:小耳/无耳畸形;耳前赘;耳前瘘管;中耳发育不良;内耳发育不良;不同程度耳聋。
- 脊椎:半椎体、块椎体、脊柱侧凸/后凸、寰椎枕化和脊柱裂,主要表现在颈椎、胸椎和肋骨。
- 头部:大头/小头畸形。
- 面部:颧骨、上颌骨或下颌区发育不良,特别是下颌支和下颌髁及颞下颌关节发育不良;嘴角侧向裂并延伸(巨口畸形);面瘫。
- 口腔:唇裂/腭裂;腮腺分泌减少或缺乏;舌结构或功能异常;软腭功能障碍;龋齿。
- 心脏:室间隔缺损;动脉导管未闭;Fallot四联症;主动脉狭窄。
- 泌尿生殖:肾异位或融合;肾发育不全;膀胱输尿管回流;输尿管肾盂连接梗阻;输尿管重复;多囊肾。
- 其他:智力障碍(13%的患者智商小于85);颈前侧有鳃裂残余;喉异常;肺发育不良/不全;食管瘘;外部血管压迫造成气管软化;脑积水;Arnold-Chiari畸形;枕部脑膨出;胼胝体发育不全;胼胝体脂肪瘤;大脑镰钙化;透明隔发育不良;颅内皮样囊肿;单发或并发桡骨和肋骨异常等。
【Branchio-Oto-Renal Syndrome,BORS(鳃-耳-肾综合征)】
BORS是一种少见的常染色体显性遗传性疾病,主要表现为听力下降、耳前瘘管、鳃裂畸形、肾发育不良等。参见科普文章《医学科普——易被疏忽的鳃-耳-肾综合征》。
【结语】
上述鳃器异常发育相关颅面综合征,常累及多系统、多器官,需要多学科多专业(包括眼科、心脏科、内分泌科、耳鼻咽喉头颈外科、口腔颌面外科、整形外科、免疫科、儿童发育及康复科、精神心理科等),共同参与、分工协作,长期、综合系列治疗和观察随访。
【参考文献】
- 刘爽, 范欣淼, 朱莹莹,等. Treacher-Collins综合征的精准诊断与治疗(临床篇)[J]. 临床耳鼻咽喉头颈外科杂志, 2018, 32(16):5.
- McDonald-McGinn Donna M,Sullivan Kathleen E,Marino Bruno,et al.22q11.2 deletion syndrome.Nature reviews. Disease primers.2015;1:15071.doi:10.1038/nrdp.2015.71
- 程琪.Pierre Robin序列征的研究进展[J].国际儿科学杂志,2010,37(6):565-567.
- 付菲, 丁明超, 田磊,等. 第一、二鳃弓综合征诊疗进展[J]. 中华口腔医学研究杂志:电子版, 2018, 12(2):5.
- 马竞,周文浩.新生儿常见小耳畸形相关综合征的遗传特征[J].中国当代儿科杂志,2022,24(6):614-619.
【附相关科普文章】
如需了解既往相关科普,可参考以往的系列文章(《先天性鳃裂畸形诊治的“变与不变”》、《六载耕耘 耳鼻喉科打造先天性鳃源性畸形治疗全国品牌》、、《鳃裂囊肿(瘘管),你找对医生了吗?》、《鳃裂畸形:小病种?or 大品牌》、《因为专注,所以专业——你可信赖的鳃裂瘘管医生(暨头颈部先天性疾病诊治的工作积累)》、《透过组织病理学的视角,看鳃裂畸形!(上篇)》、《透过组织病理学的视角,看鳃裂畸形!(下篇)》、《鳃裂畸形,可以选择“带瘘生存”吗?》、《易被疏忽的鳃-耳-肾综合征》、《鳃裂畸形手术岂可止于病灶的简单挖除?(如何避免鳃裂畸形术后复发和并发症?)》、《孕期鳃裂囊肿怎么办?》、《内外兼修,除鳃亦需防疤:鳃裂畸形术后颈面部疤痕的防治》、《鳃裂畸形的“环肥燕瘦”》、《鳃裂的“是非功过”》、《善观风色,相机而动:鳃裂畸形外科手术时机的把控》、《儿童鳃裂畸形,家长如何早发现(最值得宝妈、宝爸收藏的看图识瘘——鳃裂囊瘘)》、《医学科普——天下苦“鳃裂畸形继发感染”久矣!(鳃裂畸形继发感染的疑惑及应对)》、《鳃裂畸形炎症感染期的急性疼痛,应对有方》、《十八般兵器,哪一款能量器械是鳃裂畸形开放性外科中的“青龙偃月刀”?》、《“窦娥冤”,不是所有的颈深间隙感染都源自梨状窝瘘(写在350例梨状窝瘘诊治完成之际)!》、《在这里,梨状窝瘘“不罕见”(写在500例梨状窝瘘诊治完成之际)!》、《拿什么来拯救你,复发性梨状窝瘘(写在600例梨状窝瘘诊治完成之际)》、《道阻且长,行则将至:梨状窝瘘诊治的探索和创新之路(记“陈氏改良梨状窝瘘管切除术”获奖)》、《梨状窝瘘手术的难与易(写在700例梨状窝瘘诊治完成之际)》、《精雕细琢方为器,千锤百炼始成钢:“陈良嗣鳃裂畸形专病诊治品牌”的铸就体会(写在1500余例鳃裂畸形诊治完成之际)》、《搭上开往春天的火车,追寻暗夜里的明灯,医缘暖人心(写在1500例鳃裂畸形诊治完成之际)》、《见风是雨?莫把继发性颈部炎性皮瘘误当鳃裂瘘管(上篇)》、《见风是雨?莫把继发性颈部炎性皮瘘误当鳃裂瘘管(下篇)》、《鳃裂畸形手术切口设计有讲究:因型而异、合理规划(2024年六一儿童节专刊:宝妈宝爸最想知道的鳃裂囊瘘切口设计)》、《事缓则圆:鳃裂畸形诊治中的“慢”(写在1600余例鳃裂畸形诊治完成之际)》、《与鳃器异常发育相关的颅面综合征》等。)
End
文:陈良嗣头颈腺体、先天疾病及儿童头颈外科工作室
广东.广州
终稿于2024年11月7日(甲辰立冬.金)
(ILMSS、GBO、GBMF)
如需解惑和帮助,可识别以下二维码,网络问诊或电话咨询。

如欲了解更多的头颈先天疾病知识,可识别以下“陈良嗣医生的科普号”二维码

本文是陈良嗣版权所有,未经授权请勿转载。本文仅供健康科普使用,不能做为诊断、治疗的依据,请谨慎参阅
评论