


 三甲
三甲
46,XY性别发育异常的性腺处理
性别发育异常/差异(disorders/differences of sex development, DSD)是一种先天性染色体、性腺和表型性别的发育异常或不匹配。2006芝加哥会议将性分化异常分为3类:性染色体DSD、46,XXDSD(男性化女性)及46,XYDSD(男性化不足的男性)。其中46,XYDSD包括性腺(睾丸)发育异常、雄激素合成及作用异常、重度尿道下裂和泄殖腔外翻。目前针对46,XYDSD患儿的性腺处理仍存在一定争议,内容包括46,XYDSD患儿何时需要性腺活检及探查、46,XYDSD患儿性腺恶变风险、如何随访检测性腺恶变情况、性腺切除的指征及时机等。出于保护患儿生殖功能的考虑,46,XYDSD患儿性腺处理方案的选择也存在诸多的影响因素。本文将在复习相关文献的基础上,结合笔者临床经验从以上几个方面浅谈46,XYDSD的性腺处理。
一、性腺活检及探查
DSD患儿病因诊断对于治疗策略的选择非常重要。46,XXDSD通常可以精确诊断出病因,但46,XYDSD仅有50%左右能诊断出明确病因。具有确定意义的体格检查是发现一侧或双侧性腺,可有效排除男性化的女性患儿。少见的情况为卵睾下降至腹股沟区,如果性腺两极结构不对称,则高度怀疑为卵睾组织,可通过超声检查进一步确诊。如果在体表均不可触及双侧睾丸,则可通过检测促黄体生成素(luteinizing hormone,LH)是否显著升高来判断睾丸的存在与否;也可通过人绒毛促性腺激素(human chorionic gonadotropin, HCG)刺激实验来确定是否存在有功能的睾丸组织。除无睾症外,HCG刺激实验也可诊断5α-还原酶缺乏(HCG刺激之后睾酮/双氢睾酮升高),还可以鉴别雄激素合成障碍(对HCG刺激后无睾酮升高)和雄激素受体不敏感(HCG刺激后睾酮升高),但确诊仍需基因检测。可以通过血清苗勒氏管抑制物质和抑制素B来确定睾丸是否存在。此外,还可采用聚合酶链式反应(polymerase chain reaction,PCR)法对静脉血进行DNA检测,以精确诊断DSD患儿的基因异常(尤其可以确定雄激素受体以及酶的异常)。如果通过以上检查结果仍不能确诊,则可行腹腔探查、腹腔镜探查及性腺活检进一步诊断,其中活检需要沿性腺长轴纵向深部取材。混合性腺发育不全的条纹性腺可依据冰冻结果诊断,其他则需依据石蜡切片结果。只有获得最终病理结果及性别认定后,才能进行性腺或生殖器官的切除。
二、46,XYDSD性腺恶变风险
生殖细胞肿瘤(germ cell tumors, GCTs)癌前病变包括原位生殖细胞肿瘤(germ cell neoplasia in situ,GCNIS)、小管内生殖细胞肿瘤(intratubular germ cell neoplasia, ITGCN)和性腺母细胞瘤(gonadoblastoma,GB)。原位癌(carcinoma-in-situ,CIS)仅发生于分化很好且包含曲细精管的睾丸组织。对于各种男性化不足综合症患儿,由于其性腺分化良好,因此仅有可能发生CIS而不发生GB。GB主要见于未分化的性腺组织及原始性腺索,几乎都发生在有Y染色体物质的发育不良性腺,可在出生时存在,也可在在生后的一段时间内出现,其中50%左右将发展为恶性肿瘤。对于GB,如果出现部分睾丸分化也可发生CIS。GB包含生殖细胞、基质以及未成熟支持细胞,可有钙化。单纯GB并非恶性肿瘤,不发生转移,但可以转为恶性的生殖细胞瘤或精原细胞瘤;若GB侵及性腺基质,女性可导致无性细胞瘤,男性可导致精原细胞瘤。
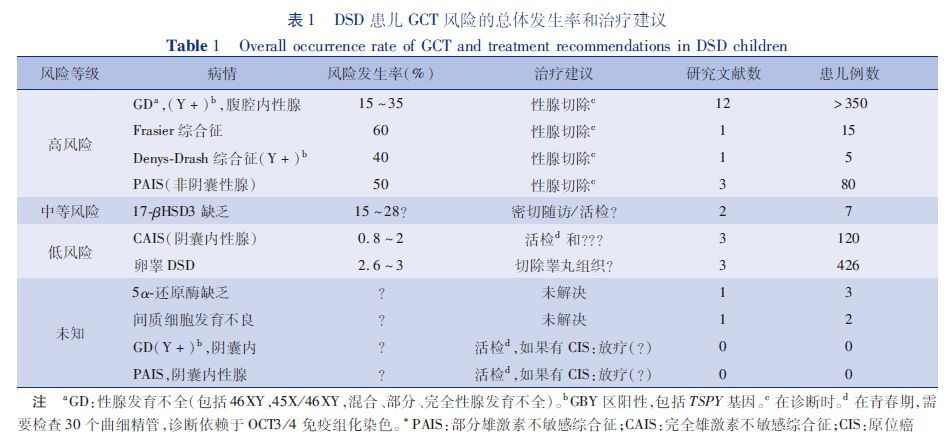
GCT肿瘤风险分层(表1):GCT风险等级是由预防性切除的性腺或活检获得的GCNIS/GB结果推测得来的,但并不清楚GCNIS/GB转化为恶性肿瘤的机率。46,XYDSD中GCT风险分层如下:①完全性腺不发育和部分性腺发育不全:46,XY完全性腺不发育性腺肿瘤发生率为15%~50%,而46,XY部分性腺发育不全性腺肿瘤发生率为16%~30%。Deny-drash综合征和Frasier综合征表现为性腺发育不全和肾脏病变,这两种综合征的发生是由于Wilms’肿瘤抑制基因(Wilms’ tumor suppressor gene,WT1)发生突变而引起,其中合并WT1基因突变的46,XY性腺发育不全患儿性腺肿瘤发生率可达40%~60%。部分和完全性腺发育不全最易发生的常见性腺肿瘤为GB。尽管肿瘤常在青春期后发生,但是有些性腺发育不全患儿在婴儿期就可出现性腺肿瘤。②卵睾DSD:在卵睾DSD中生殖细胞肿瘤发生取决于性腺分化程度及是否存在Y染色体物质。然而46,XY卵睾性腺恶变风险率较低,为2%~10%。③睾酮合成障碍性腺恶变率低(<5%)。性别发育异常共识中报道17β-HSD3(17β-hydroxyteroid dehydrogenase)缺乏肿瘤发生率高达28%,但是该数据仅基于7例17β-HSD3缺乏的患儿,且其中有2例出现了睾丸肿瘤。④原来认为完全雄激素受体不敏感的恶变率为9%~22%,最近报道的结果为0.8%~2%,仅仅比隐睾稍高。最常见的肿瘤为精原细胞瘤、性腺母细胞瘤,小管内生殖细胞瘤和支持细胞瘤。⑤部分雄激素受体不敏感比完全雄激素受体不敏感性腺肿瘤的发生率高,达15%,隐睾未治疗者患GCT风险高达50%,而部分雄激素受体不敏感阴囊内睾丸者患肿瘤风险尚不清楚,但还有发生乳腺癌的风险。
三、随访检测性腺恶变
腹腔性腺需要移出腹腔,放到腹股沟(最好是阴囊)内,以便于监测恶变。如不能移出腹腔,则需切除性腺。目前尚无可信且特异度较高的方法检测有恶变风险的性腺恶变发生。建议DSD阴囊内睾丸自青春期开始后每月进行自检。阴囊内或腹股沟性腺可通过超声检查,建议从青春后期开始每年超声随访性腺状况。尽管不能通过超声诊断原位癌,但可检测到睾丸实质回声不规则及微结石的存在(但二者对于CIS并无特异诊断价值)。不建议应用超声、MRI或CT对位于腹腔的发育不良性腺进行检测。性腺活检结果虽然特异度较高,但较容易遗漏性腺肿瘤。
除了影像学检查及性腺活检,还有一些肿瘤标志物可以提示性腺恶变。目前临床应用的肿瘤标志物除HCG和甲胎蛋白(alpha-fetoprotein,AFP)外,还包括八聚物-结合转录因子OCT3/4(octamer-binding transcription factor OCT3/4)、Y染色体上的睾丸特异蛋白TSPY(testis-specific protein on Y)和WT-1。OCT3/4为核转录因子,在人类胚胎和干细胞中表达,是CIS、GB、GCT的病理标志物,成熟细胞中无OCT3/4表达。由于OCT3/4还可作为胚胎标志物,因此可在孕晚期及生后早期检测到,DSD患儿6个月后外观正常的生殖细胞也可表达OCT3/4,为成熟延迟的表现。为区分成熟延迟和癌前病变,可检测干细胞因子(stem cell factor SCF)和c-KIT配体(c-KIT ligand KITLG),后者为早期生殖细胞恶变的一个特异性标志物,仅在性腺ITGCN或GB中可检测到。DSD患儿发生GB与Y染色体GBY区的存在(gonadoblastoma region on Y chromosome)高度相关,TSPY的激活和表达可导致CIS/GB的发生。
四、性腺切除指征与时机
性别选择后,需要切除与选择性别不一致的性腺。此外,需要切除存在Y染色体物质的条纹性腺(46,XY完全性腺发育不全、混合性腺发育不全),以防止恶变的发生。按女性抚养的雄激素合成障碍的患儿必须在青春期前切除睾丸,以防止发生男性化。对于完全雄激素不敏感的患儿,由于睾丸可产生雌二醇,而雌二醇可导致女性化表型的转变,可保留睾丸在原位直至青春期结束以利于骨骼及乳腺的发育。如果是部分雄激素不敏感,青春期时可发生男性化,因此一经诊断即可行性腺切除。如果是卵睾,切除与选择性别不一致的性腺部分需要根据术中冰冻结果确认后,再行完全切除,术后可以通过HCG/人绝经期促性腺激素(human menopausal gonadotropin,HMG)刺激实验来确定睾丸或卵巢组织是否切除完全。对于睾丸和卵巢分界不清者,建议切除整个性腺。位于阴囊的发育不良睾丸有恶变的风险,因此建议在青春期时行睾丸活检,如果存在原位癌或小管内生殖细胞瘤,建议保留睾丸并行低剂量放疗。
关于睾丸切除的时间(青春期前还是青春期后)仍存在争议,很大程度上取决于预期的恶变风险。由于肿瘤多发生在青春期后,一些人主张延迟手术,但前提是可以安全监测性腺状况。对于合并的腹股沟疝或存在性腺相关心理问题的患儿则更多主张青春期前接受手术。此外,青春期开始后可能会出现与选择性别不一致的男性化或女性化者,为避免此类现象发生,青春期前切除性腺可能是更好的选择。对于完全性腺发育不全或部分性腺发育不全者,如果腹腔内性腺不能下降至易于监测的部位时需行切除。完全雄激素受体不敏感恶变风险低,青春期后延迟切除性腺已经被广泛接受,但有一些成年女性不同意切除性腺,则需要将腹腔性腺放置在一个更表浅的位置。睾酮合成障碍性腺恶变率较低(1%~15%),但对于46,XYDSD睾酮合成障碍完全女性外生殖器且选择女性性别者,仍建议儿童期切除睾丸以防止恶变。对于任何未成年人性腺切除手术,除非有健康风险(如发育不良性腺的恶变风险)或通过治疗也不可能具有生育功能的情况,其他情况的处理应该慎重。
对于性腺切除的决定应该做到个体化,并充分考虑社会心理因素。目前已有报道的程序化风险因素(包括年龄、性腺位置、TSPY阳性检测结果、外生殖器表型)还未能被广泛接受用于临床决策。随着越来越多与DSD病因相关的基因被识别,可能会逐步衍生出恶性GCT筛查和预防的基因特异性诊断方法。预防性性腺切除的利弊分析需要综合考虑可供选择的筛查手段及其成功率,同时能够基本确定对应的早期治疗的策略。对于保留性腺的DSD患儿,影像学监测腹腔内性腺GCT是不可靠的,可将性腺放置在活检时腔镜Trocar部位或腹股沟,以便检查。
五、出于生殖能力考虑的性腺处理方案选择
以前我们认为,几乎所有的46,XYDSD患儿都没有生育功能,但有学者研究发现卵睾DSD,Denys-Drash综合征等DSD患儿的性腺中存在生殖细胞。所以46,XYDSD患儿性腺处理也应尽可能保留患儿生育功能。不同类型 DSD患儿性腺处理方案选择如下:①46,XY完全(单纯)性腺发育不全(Swyer综合征):此类患儿没有生育功能,因双侧性腺恶变风险高,需要切除双侧条纹性腺;如按女性抚养,可通过赠卵完成生育。②46,XY混合性腺发育不全:不论选择男性还是女性,此类患儿通常不育。如果选择男性性别,可行睾丸下降固定术,需要在严密监测性腺肿瘤发生(定期体检及超声检查)及预防性性腺切除加雄激素替代两种方案间进行选择。③46,XY卵睾DSD:卵睾的卵巢组织发育的更正常一些,而睾丸组织常发育不良,很少有正常的生殖细胞。如果内生殖器正常,按女性抚养有保留生育能力的可能。按男性抚养,通过取精和细胞内精子注射技术也有成功生育的报道。如果按女性抚养,所有睾丸和午非氏管结构都需要切除,如果是卵睾,则需切除睾丸部分,术后可以通过HCG刺激实验确定睾丸组织是否切除完全。对于睾丸和卵巢分界不清者,建议切除性腺。尽管可能需要激素治疗替代,但保留的卵巢组织在青春期可能有正常的卵巢功能。对于按女性抚养者也需要监测性腺发生肿瘤的风险。如果选择男性性别,需切除所有卵巢和苗勒氏管结构,由于性腺恶变风险高且没有生育可能,需要考虑青春期性腺切除激素替代,如果保留性腺,至少需要长期超声监测性腺恶变风险。对于阴囊内的发育不良性腺建议青春期活检,如果有睾丸CIS,建议冷冻精子并行低剂量放疗。无论选择哪种性别,青春期根据残余性腺组织的功能确定是否需要行激素替代治疗。④17β-羟类固醇氧化还原酶(3型)缺乏:目前为止,17β-羟类固醇氧化还原酶对于男性性别认定的证据支持等级略低于5α还原酶,且17β-HSD3有中等风险发生生殖细胞肿瘤,并且至今无生育个案的报道。因此,在合适的情况下也可以选择女性性别,如一直按女性抚养,青春期前诊断可以选择切除睾丸,或者阻断青春期直至患儿自己做出决定。如保留睾丸则需监测恶变。⑤完全雄激素受体不敏感:大脑无雄激素化,典型的女性性别认定、性行为及性取向。外生殖器不需接受整形手术,性腺切除后需激素替代治疗。⑥部分雄激素受体不敏感按女性抚养者的性别焦虑高于男性抚养者(20% vs. 7%)。但大多数部分雄激素受体不敏感患儿的性别身份和性别认定一致。部分雄激素受体不敏感患儿对生殖器重建及性生活质量不满意的情况很常见。如果阴茎对睾酮的反应好可选择男性,如睾酮治疗无效则可选择女性。⑦5α还原酶缺乏:由于出生时为女性外生殖器外观,云南菜青春期前大多按女性抚养,青春期发生男性化后56%~63%患儿性别身份从女性转为男性,且选择男性性别可能保留生育能力,因此通常建议选择男性性别。如保留睾丸,青春期通常不需激素替代治疗。⑧苗勒氏管永存综合征:患儿均为男性表型,性别认定为男性,需行睾丸固定术。苗勒氏管永存综合征患儿存在长期隐睾可导致生殖细胞成熟异常、曲细精管萎缩、间质纤维化和严重的间质细胞发育不良,生精细胞从精原细胞成熟为初级精母细胞的过程可能发生障碍;此外,还可能会出现附睾和输精管异常导致的梗阻性无精。苗勒氏管永存综合征患儿常需依赖辅助生殖技术(associated reproduction technique, ART)保留生育能力。
总之,对于46,XYDSD患儿性腺处理的决定应该个体化,需综合考虑性腺恶变风险、生殖潜能以及最重要的性心理认定。如果内分泌检查不能确诊,可进行腹腔探查、腹腔镜探查及性腺活检进一步判断。腹腔性腺需移出腹腔放到腹股沟(最好是阴囊内)以便监测恶变,如不能移出腹腔则需切除性腺。目前尚无可信且特异的方法检测有恶变风险的性腺恶变发生。由于46,XYDSD睾丸存在一定功能,因此更多学者建议选择男性性别,而对性腺的处理通常应与所选性别一致。现在更多学者倡导患儿自己决定性别选择。目前建议性腺切除的指征:①(早期)生殖细胞癌;②(预期)性腺分泌激素有反向作用;③患儿自检或影像监测性腺恶变的依从性差,且患儿要求切除性腺;④存在Y染色体物质的条纹性腺(包括完全性腺发育不全、混合性腺发育不全)。而睾丸切除的最佳时间(青春期前还是青春期后)仍存争议,主要依赖于预期恶变的风险。
文献来源:杨屹,殷晓鸣.46,XY性别发育异常的性腺处理.临床小儿外科杂志,2019,18(03):167-171.
本文为转载文章,如有侵权请联系作者删除。本文仅供健康科普使用,不能做为诊断、治疗的依据,请谨慎参阅
评论